 RUXIENCE 100 MG CONCENTRADO PARA SOLUCION PARA PERFUSION
RUXIENCE 100 MG CONCENTRADO PARA SOLUCION PARA PERFUSION

| ATC: Rituximab |
| PA: Rituximab |
Envases
- Env. con 1 vial de 10 ml
- H: Medicamento de uso hospitalario
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica Medicamento Biosimilar
Medicamento Biosimilar- No sustit.: Medicamento NO sustituible por el farmacéutico (Biológicos)
- Fi: Medicamento incluido en la financiación del SNS
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 728339
- EAN13: 8470007283393
- Conservar en frío: Sí
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
RUXIENCE 100 mg Concent. para sol. para perfus.
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Ruxience 100 mg concentrado para solución para perfusión
Cada ml contiene 10 mg de rituximab.
Cada vial de 10 ml contiene 100 mg de rituximab.
Ruxience 500 mg concentrado para solución para perfusión
Cada ml contiene 10 mg de rituximab.
Cada vial de 50 ml contiene 500 mg de rituximab.
Rituximab es un anticuerpo monoclonal quimérico de ratón/humano modificado genéticamente que representa una inmunoglobulina glucosilada con las regiones constantes de la IgG1 humana y secuencias de la región variable de las cadenas ligeras y pesadas murinas. El anticuerpo se produce mediante cultivo en suspensión de células de mamífero (ovario de hámster chino) y se purifica mediante cromatografía de afinidad y de intercambio iónico, incluidos los procedimientos específicos de inactivación y eliminación viral.
Excipiente con efecto conocido
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis.
Para consultar la lista completa de excipientes, ver sección 6.1
3. - FORMA FARMACÉUTICA
Concentrado para solución para perfusión (concentrado estéril).
Líquido de incoloro a marrón amarillento claro y de transparente a ligeramente opalescente.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de RUXIENCE 100 mg Concent. para sol. para perfus.
Ruxience está indicado en adultos para las siguientes indicaciones:
Linfoma no Hodgkin (LNH)
Ruxience está indicado para el tratamiento en combinación con quimioterapia de pacientes adultos con linfoma folicular en estadio III-IV no tratados previamente.
El tratamiento de mantenimiento con Ruxience está indicado para el tratamiento de pacientes adultos con linfoma folicular que hayan respondido al tratamiento de inducción.
Ruxience en monoterapia está indicado para el tratamiento de pacientes adultos con linfoma folicular en estadio III-IV que son quimioresistentes o están en su segunda o posterior recidiva tras la quimioterapia.
Ruxience está indicado en combinación con quimioterapia CHOP (ciclofosfamida, doxorrubicina, vincristina, prednisolona) para el tratamiento de pacientes adultos con linfoma no Hodgkin difuso de células B grandes CD20 positivo.
Ruxience está indicado en combinación con quimioterapia en el tratamiento de pacientes pediátricos (de edad ≥ 6 meses a < 18 años) con linfoma difuso de células B grandes CD20 positivo (LBDCG), linfoma de Burkitt (LB)/leucemia de Burkitt (leucemia aguda de células B maduras) (LLA-B madura) o linfoma similar a Burkitt (LBL) en estadios avanzados no tratados previamente.
Leucemia linfocítica crónica (LLC)
Ruxience en combinación con quimioterapia está indicado para el tratamiento de pacientes con LLC no tratada previamente y recidivante/refractaria. Solo se dispone de datos limitados sobre la eficacia y la seguridad de los pacientes tratados previamente con anticuerpos monoclonales, incluido rituximab, o pacientes refractarios al tratamiento previo con rituximab y quimioterapia.
Ver sección 5.1 para más información.
Artritis reumatoide
Ruxience en combinación con metotrexato está indicado para el tratamiento de pacientes adultos con artritis reumatoide activa grave que hayan tenido una respuesta inadecuada o intolerancia a otros fármacos antirreumáticos modificadores de la enfermedad (FARME) incluyendo uno o más tratamientos inhibidores del factor de necrosis tumoral (TNF).
Se ha demostrado que Ruxience reduce la tasa de progresión del daño articular medido por Rayos X y mejora la función física, cuando se administra en combinación con metotrexato.
Granulomatosis con poliangeitis y poliangeitis microscópica
Ruxience, en combinación con glucocorticoides, está indicado para el tratamiento de pacientes adultos con granulomatosis con poliangeitis (granulomatosis de Wegener) (GPA) y poliangeitis microscópica (PAM) activas y graves.
Ruxience, en combinación con glucocorticoides, está indicado para la inducción de la remisión de pacientes pediátricos (de edad ≥ 2 a < 18 años) con GPA (Wegener) y con PAM, activas y graves.
Pénfigo vulgar
Ruxience está indicado para el tratamiento de pacientes con pénfigo vulgar (PV) moderado o grave.
4.2 - Posología y administración de RUXIENCE 100 mg Concent. para sol. para perfus.
4.3 - Contraindicaciones de RUXIENCE 100 mg Concent. para sol. para perfus.
Contraindicaciones para el uso en el linfoma no Hodgkin y la leucemia linfocítica crónica
Hipersensibilidad al principio activo o a las proteínas de ratón, o a alguno de los excipientes incluidos en la sección 6.1.
Infecciones graves y activas (ver sección 4.4).
Pacientes gravemente inmunodeprimidos.
Contraindicaciones para el uso en artritis reumatoide, granulomatosis con poliangeitis, poliangeitis microscópica y pénfigo vulgar
Hipersensibilidad al principio activo o a las proteínas de ratón, o a alguno de los excipientes incluidos en la sección 6.1.
Infecciones graves y activas (ver sección 4.4).
Pacientes gravemente inmunodeprimidos.
Insuficiencia cardíaca grave (clase IV de la Asociación de Cardiología de Nueva York) o enfermedad cardíaca grave no controlada (ver sección 4.4 sobre otras enfermedades cardiovasculares).
4.4 - Advertencias y Precauciones de RUXIENCE 100 mg Concent. para sol. para perfus.
Trazabilidad
Con objeto de mejorar la trazabilidad de los medicamentos biológicos, el nombre y el número de lote del medicamento administrado deben estar claramente registrados.
Leucoencefalopatía multifocal progresiva
Todos los pacientes tratados con Ruxience para artritis reumatoide, GPA, PAM o pénfigo vulgar deben recibir la tarjeta de información al paciente con cada perfusión. La tarjeta de información contiene información de seguridad importante para los pacientes sobre el potencial aumento del riesgo de sufrir infecciones, incluida la leucoencefalopatía multifocal progresiva (LMP).
Se han notificado casos muy raros de LMP mortal tras el uso de rituximab. Se debe vigilar a los pacientes a intervalos regulares para detectar cualquier síntoma neurológico nuevo o que haya empeorado o signos que puedan indicar LMP. Si se sospecha de LMP, se deben suspender las dosis adicionales hasta que se haya descartado la LMP. El médico debe evaluar al paciente para determinar si los síntomas son indicativos de disfunción neurológica y, de ser así, si estos síntomas son indicativos de LMP. La consulta con un neurólogo se debe considerar clínicamente indicada.
Si existe alguna duda, se debe considerar realizar una evaluación adicional, incluyendo una resonancia magnética, preferiblemente con contraste, el análisis del líquido cefalorraquídeo (LCR) para detectar ADN del virus JC y repetir evaluaciones neurológicas.
El médico debe estar especialmente alerta frente a los síntomas indicativos de LMP que el paciente pueda advertir (por ejemplo, síntomas cognitivos, neurológicos o psiquiátricos). También se debe aconsejar a los pacientes que informen a su pareja o cuidadores sobre su tratamiento, ya que ellos pueden notar síntomas de los que el paciente no es consciente.
Si un paciente desarrolla LMP, el tratamiento con Ruxience se debe suspender permanentemente.
Se ha observado una estabilización o mejoría del desenlace clínico después de la reconstitución del sistema inmunitario en pacientes inmunodeprimidos con LMP. Aún se desconoce si la detección temprana de LMP y la suspensión del tratamiento con rituximab pueden conducir a una estabilización similar o a una mejoría del desenlace clínico.
Linfoma no Hodgkin y leucemia linfocítica crónica
Reacciones relacionadas con la perfusión
El uso de rituximab está asociado a reacciones relacionadas con la perfusión, que pueden estar relacionadas con la liberación de citoquinas y/u otros mediadores químicos. El síndrome de liberación de citoquinas puede ser clínicamente indistinguible de las reacciones de hipersensibilidad aguda.
Este conjunto de reacciones que incluye el síndrome de liberación de citoquinas, el síndrome de lisis tumoral y las reacciones anafilácticas y de hipersensibilidad se describen a continuación.
Se han notificado reacciones graves relacionadas con la perfusión con resultados mortales durante el uso post comercialización de la formulación intravenosa de rituximab, con una aparición que oscila entre 30 minutos y 2 horas después de comenzar la primera perfusión intravenosa de rituximab. Estas se caracterizaron por acontecimientos pulmonares y en algunos casos incluyeron lisis tumoral rápida y características del síndrome de lisis tumoral además de fiebre, escalofríos, rigidez, hipotensión arterial, urticaria, angioedema y otros síntomas (ver sección 4.8).
El síndrome de liberación de citoquinas grave se caracteriza por una disnea grave, a menudo acompañada de broncoespasmo e hipoxia, además de fiebre, escalofríos, rigidez, urticaria y angioedema. Este síndrome puede estar relacionado con algunas características del síndrome de lisis tumoral, como hiperuricemia, hiperpotasemia, hipocalcemia, hiperfosfatemia, fallo renal agudo y aumento de la lactato deshidrogenasa (LDH) y puede estar relacionado con fallo respiratorio agudo y muerte. El fallo respiratorio agudo puede estar acompañado de acontecimientos tales como infiltración intersticial pulmonar o edema, visible en una radiografía de tórax. El síndrome con frecuencia se manifiesta durante la primera o segunda hora después de comenzar la primera perfusión. Los pacientes con antecedentes de insuficiencia pulmonar o aquellos con infiltración tumoral pulmonar pueden tener un mayor riesgo de resultados deficientes y deben aumentar las precauciones durante el tratamiento. Los pacientes que desarrollan síndrome de liberación de citoquinas grave deben interrumpir la perfusión inmediatamente (ver sección 4.2) y deben recibir un tratamiento sintomático intensivo. Dado que a la mejoría inicial de los síntomas clínicos puede seguir un empeoramiento, estos pacientes se deben monitorizar estrechamente hasta que el síndrome de lisis tumoral y la infiltración pulmonar se hayan resuelto o descartado. Una vez resueltos completamente los signos y síntomas, raramente se repite el síndrome de liberación de citoquinas grave en tratamientos posteriores.
Los pacientes con una carga tumoral elevada o con un número elevado de células malignas circulantes (≥ 25 x 109/l), como los pacientes con LLC, que pueden tener un riesgo mayor de síndrome de liberación de citoquinas especialmente grave, se deben tratar con extrema precaución. Se debe vigilar estrechamente a esos pacientes durante la primera perfusión. En estos pacientes se debe considerar el uso de una velocidad de perfusión reducida en la primera perfusión o el fraccionamiento de la dosis durante dos días durante el primer ciclo y cualquier ciclo posterior si el recuento de linfocitos aún es > 25 x 109/l.
Se han observado reacciones adversas de todo tipo relacionadas con la perfusión en el 77 % de los pacientes tratados con rituximab (incluido el síndrome de liberación de citoquinas acompañado de hipotensión arterial y broncoespasmo en el 10 % de los pacientes) (ver sección 4.8). Estos síntomas generalmente son reversibles con la interrupción de la perfusión de rituximab y la administración de un antipirético, un antihistamínico y, ocasionalmente, oxígeno, solución salina intravenosa o broncodilatadores y, si es necesario, glucocorticoides. Ver síndrome de liberación de citoquinas más arriba para consultar las reacciones graves.
Se han notificado reacciones anafilácticas y de hipersensibilidad tras la administración intravenosa de proteínas a los pacientes. A diferencia del síndrome de liberación de citoquinas, las reacciones de hipersensibilidad verdaderas aparecen generalmente minutos después de comenzar la perfusión. Conviene disponer de medicamentos para el tratamiento de las reacciones de hipersensibilidad, por ejemplo, epinefrina (adrenalina), antihistamínicos y glucocorticoides para uso inmediato si ocurre una reacción alérgica durante la administración de rituximab. Las manifestaciones clínicas de la anafilaxia pueden parecer similares a las manifestaciones clínicas del síndrome de liberación de citoquinas (descrito más arriba). Las reacciones atribuidas a la hipersensibilidad se han notificado con menos frecuencia que las atribuidas a la liberación de citoquinas.
Otras reacciones notificadas en algunos casos fueron infarto de miocardio, fibrilación auricular, edema pulmonar y trombocitopenia reversible aguda.
Dado que puede aparecer hipotensión arterial durante la administración de rituximab, se debe considerar la interrupción de la administración de antihipertensores durante 12 horas antes de la perfusión de Ruxience.
Trastornos cardíacos
Se han notificado casos de angina de pecho, arritmias cardíacas tales como flutter y fibrilación auricular, fallo cardíaco y/o infarto de miocardio en pacientes tratados con rituximab. Por lo tanto, se debe vigilar estrechamente a los pacientes con antecedentes de enfermedad cardíaca y/o cardiotoxicidad asociada a quimioterapia.
Toxicidades hematológicas
Aunque rituximab no es mielosupresor en monoterapia, se debe tener precaución al considerar el tratamiento de pacientes con un recuento de neutrófilos < 1,5 x 109/l y/o recuento de plaquetas < 75 x 109/l, ya que la experiencia clínica en esta población es limitada. Rituximab se ha utilizado en 21 pacientes que se sometieron a un trasplante autólogo de médula ósea y otros grupos de riesgo con una función presumiblemente reducida de la médula ósea sin inducir mielotoxicidad.
Se deben realizar hemogramas regulares, incluidos recuentos de neutrófilos y plaquetas, durante el tratamiento con Ruxience.
Infecciones
Pueden aparecer infecciones graves, e incluso mortales, durante el tratamiento con rituximab (ver sección 4.8). Ruxience no se debe administrar a pacientes con una infección activa y grave (por ejemplo, tuberculosis, sepsis e infecciones oportunistas, ver sección 4.3).
Los médicos deben tener precaución al considerar el uso de Ruxience en pacientes con antecedentes de infecciones recurrentes o crónicas o con afecciones subyacentes que pueden predisponer aún más a los pacientes a infecciones graves (ver sección 4.8).
Se han notificado casos de reactivación de la hepatitis B en sujetos que recibieron rituximab, incluida hepatitis fulminante con desenlace mortal. La mayoría de estos sujetos también estuvieron expuestos a quimioterapia citotóxica. La información limitada de un estudio en pacientes con LLC recidivante/refractaria sugiere que el tratamiento con rituximab también puede empeorar el resultado de las infecciones primarias por hepatitis B. Se debe investigar la presencia del virus de la hepatitis B (VHB) en todos los pacientes antes de iniciar el tratamiento con Ruxience. Como mínimo, esto debe incluir el estado del HBsAg y del HBcAb. Estos se pueden complementar con otros marcadores apropiados según las guías locales. No se debe tratar con Ruxience a los pacientes con hepatitis B activa. Los pacientes con serología positiva de hepatitis B (HBsAg o HBcAb) deben consultar a expertos en enfermedades hepáticas antes de comenzar el tratamiento y deben ser vigilados y tratados siguiendo los estándares médicos locales para prevenir la reactivación de la hepatitis B.
Se han notificado casos muy raros de leucoencefalopatía multifocal progresiva (LMP) durante el uso post-comercialización de rituximab en LNH y LLC (ver sección 4.8). La mayoría de los pacientes habían recibido rituximab en combinación con quimioterapia o como parte de un trasplante de células madre hematopoyéticas.
Se han notificado casos de meningoencefalitis enteroviral incluyendo casos mortales después del uso de rituximab.
Falsos negativos en las pruebas serológicas para infecciones
Debido al riesgo de falsos negativos en las pruebas serológicas para infecciones, se deben considerar herramientas diagnósticas alternativas en el caso de pacientes que presenten síntomas indicativos de enfermedad infecciosa rara, p. ej. Fiebre del Nilo occidental o neuroborreliosis.
Inmunización
La seguridad de la inmunización con vacunas con virus vivos, después del tratamiento con rituximab, no se ha estudiado para pacientes con LNH y LLC y no se recomienda la vacunación con vacunas con virus vivos. Los pacientes tratados con Ruxience pueden recibir vacunas inactivadas; sin embargo, con las vacunas inactivadas las tasas de respuesta se puede reducir . En un estudio no aleatorizado, los pacientes adultos con LNH recidivante de bajo grado que recibieron rituximab en monoterapia en comparación con controles sanos no tratados tuvieron una menor tasa de respuesta a la vacunación con antígeno de recuerdo del tétanos (16 % frente al 81 %) y neoantígeno Keyhole Limpet Haemocyanin (KLH) (4 % frente al 76 % cuando se evaluó un aumento de > 2 veces en el título de anticuerpos). Se pueden asumir resultados similares para los pacientes con LLC teniendo en cuenta las similitudes entre ambas enfermedades, aunque no se ha investigado en ensayos clínicos.
Los títulos de anticuerpos medios antes del tratamiento frente a un panel de antígenos (Streptococcus pneumoniae, gripe A, paperas, rubéola, varicela) se mantuvieron durante al menos 6 meses después del tratamiento con rituximab.
Reacciones cutáneas
Se han notificado reacciones cutáneas graves, como necrólisis epidérmica tóxica (síndrome de Lyell) y síndrome de Stevens-Johnson, algunas con desenlace mortal (ver sección 4.8). En caso de que aparezcan tales acontecimientos con una sospecha de relación con rituximab, el tratamiento se debe suspender permanentemente.
Población pediátrica
Se dispone solo de datos limitados para pacientes menores de 3 años. Para más información consulte la sección 5.1.
Artritis reumatoide, granulomatosis con poliangeitis (GPA), poliangeitis microscópica (PAM) y pénfigo vulgar
Poblaciones con artritis reumatoide no tratadas previamente con metotrexato (MTX)
No se recomienda el uso de rituximab en pacientes no tratados previamente con MTX ya que no se ha establecido una relación favorable de beneficio-riesgo.
Reacciones relacionadas con la perfusión
Rituximab está asociado con reacciones relacionadas con la perfusión (RRP), que pueden estar relacionadas con la liberación de citoquinas y/u otros mediadores químicos.
Se han notificado RRP graves con desenlace mortal en pacientes con artritis reumatoide durante el periodo de post-comercialización. En artritis reumatoide, la mayoría de los acontecimientos relacionados con la perfusión notificados en los ensayos clínicos fueron de gravedad leve a moderada. Los síntomas más frecuentes fueron reacciones alérgicas como dolor de cabeza, prurito, irritación de garganta, rubor, erupción cutánea, urticaria, hipertensión arterial y pirexia. En general, la proporción de pacientes que experimentaron cualquier reacción a la perfusión fue mayor después de la primera perfusión que después de la segunda perfusión de cualquier ciclo de tratamiento. La frecuencia de las RRP disminuyó con los ciclos posteriores (ver sección 4.8). Las reacciones notificadas fueron generalmente reversibles tras la reducción de la velocidad o interrupción de la perfusión de rituximab y la administración de un antipirético, un antihistamínico y, de vez en cuando, oxígeno, solución salina intravenosa o broncodilatadores y, en caso de necesidad, glucocorticoides. Se debe vigilar estrechamente a los pacientes con afecciones cardíacas preexistentes y a aquellos que hayan experimentado reacciones adversas cardiopulmonares previas. Según la gravedad de la RRP y de las intervenciones requeridas se suspenderá Ruxience de forma temporal o permanente. En la mayoría de los casos, la perfusión se puede reanudar al 50 % de la velocidad anterior (por ejemplo, de 100 mg/hora a 50 mg/hora) cuando los síntomas se hayan resuelto por completo.
Deben estar disponibles para uso inmediato medicamentos para el tratamiento de las reacciones de hipersensibilidad, por ejemplo, epinefrina (adrenalina), antihistamínicos y glucocorticoides para su uso inmediato si ocurre una reacción alérgica durante la administración de Ruxience.
No existen datos sobre la seguridad de rituximab en pacientes con insuficiencia cardíaca moderada (clase III de la Asociación de Cardiología de Nueva York) o enfermedad cardiovascular grave no controlada. En pacientes tratados con rituximab, se ha observado la aparición de afecciones cardíacas isquémicas preexistentes que se vuelven sintomáticas, como la angina de pecho, fibrilación y flutter. Por lo tanto, en pacientes con antecedentes cardíacos conocidos y aquellos que han experimentado reacciones adversas cardiopulmonares previas, se debe considerar el riesgo de complicaciones cardiovasculares, resultantes de las reacciones a la perfusión, antes del tratamiento con Ruxience y se debe vigilar estrechamente a los pacientes durante la administración. Dado que puede aparecer hipotensión arterial durante la perfusión de rituximab, se debe considerar la interrupción de la administración de antihipertensores durante 12 horas antes de la perfusión de Ruxience.
Las RRP en pacientes con GPA, PAM y pénfigo vulgar concordaron con las observadas en pacientes con artritis reumatoide en los ensayos clínicos y tras la comercialización (ver sección 4.8).
Trastornos cardíacos
Se han notificado casos de angina de pecho, arritmias cardíacas como flutter y fibrilación auricular, insuficiencia cardíaca y/o infarto de miocardio en pacientes tratados con rituximab. Por lo tanto, se debe vigilar estrechamente a los pacientes con antecedentes de enfermedad cardíaca (ver “Reacciones relacionadas con la perfusión” más arriba).
Infecciones
Teniendo en cuenta el mecanismo de acción de rituximab y el hecho de que las células B desempeñan una función importante en el mantenimiento de la respuesta inmunitaria normal, los pacientes tienen un mayor riesgo de infección después del tratamiento con rituximab (ver sección 5.1). Pueden aparecer infecciones graves, e incluso mortales, durante el tratamiento con rituximab (ver sección 4.8). Ruxience no se debe administrar a pacientes con una infección activa y grave (por ejemplo, tuberculosis, sepsis e infecciones oportunistas, ver sección 4.3) o pacientes gravemente inmunodeprimidos (por ejemplo, en los que los niveles de CD4 o CD8 son muy bajos). Los médicos deben tener precaución al considerar el uso de rituximab en pacientes con antecedentes de infecciones recurrentes o crónicas o con afecciones subyacentes que pueden predisponer aún más a los pacientes a infecciones graves, por ejemplo, hipogammaglobulinemia (ver sección 4.8). Se recomienda determinar los niveles de inmunoglobulinas antes de iniciar el tratamiento con Ruxience.
A los pacientes que notifican signos y síntomas de infección después del tratamiento con Ruxience se les debe evaluar inmediatamente y tratar de manera adecuada. Antes de administrar un ciclo siguiente de tratamiento con Ruxience, se debe reevaluar a los pacientes para detectar cualquier riesgo potencial de infecciones.
Se han notificado casos muy raros de leucoencefalopatía multifocal progresiva (LMP) mortal después del uso de rituximab para el tratamiento de la artritis reumatoide y enfermedades autoinmunitarias, incluido el lupus eritematoso sistémico (LES) y la vasculitis.
Se han notificado casos de meningoencefalitis enteroviral incluyendo casos mortales después del uso de rituximab.
Falsos negativos en las pruebas serológicas para infecciones
Debido al riesgo de falsos negativos en las pruebas serológicas para infecciones, se deben considerar herramientas diagnósticas alternativas en el caso de pacientes que presenten síntomas indicativos de enfermedad infecciosa rara, p. ej. Fiebre del Nilo occidental o neuroborreliosis.
Infecciones por el virus de la hepatitis B
Se han notificado casos de reactivación de la hepatitis B, incluidos aquellos con un desenlace mortal, en pacientes con artritis reumatoide, GPA y PAM que reciben rituximab.
Se debe investigar la presencia del virus de la hepatitis B (VHB) en todos los pacientes antes de iniciar el tratamiento con Ruxience. Como mínimo, esto debe incluir el estado del HBsAg y del HBcAb. Estos se pueden complementar con otros marcadores apropiados según las guías locales. Los pacientes con hepatitis B activa no se deben tratar con rituximab. Los pacientes con serología positiva de hepatitis B (HBsAg o HBcAb) deben consultar a expertos en enfermedades hepáticas antes de comenzar el tratamiento y deben ser vigilados y tratados siguiendo los estándares médicos locales para prevenir la reactivación de la hepatitis B.
Neutropenia tardía
Se deben determinar los neutrófilos sanguíneos antes de cada ciclo de Ruxience y regularmente hasta 6 meses después de la interrupción del tratamiento, y en presencia de signos o síntomas de infección (ver sección 4.8).
Reacciones cutáneas
Se han notificado reacciones cutáneas graves, como necrólisis epidérmica tóxica (síndrome de Lyell) y síndrome de Stevens-Johnson, algunas con desenlace mortal (ver sección 4.8). En caso de que aparezcan tales acontecimientos con una sospecha de relación con Ruxience, el tratamiento se debe suspender permanentemente.
Inmunización
Los médicos deben revisar el estado de vacunación del paciente y los pacientes, si es posible, deben estar al día con todas las inmunizaciones de acuerdo a las guías de vacunación actuales antes de iniciar el tratamiento con Ruxience. La vacunación se debe terminar al menos 4 semanas antes de la primera administración de Ruxience.
No se ha estudiado la seguridad de la inmunización con vacunas de virus vivos después del tratamiento con rituximab. Por lo tanto, no se recomienda la vacunación con vacunas de virus vivos durante el tratamiento con Ruxience o mientras haya disminución de los células B periféricas.
Los pacientes tratados con Ruxience pueden recibir vacunas inactivadas; sin embargo, las tasas de respuesta pueden reducirse con las vacunas inactivadas. En un ensayo aleatorizado, los pacientes con artritis reumatoide tratados con rituximab y metotrexato tuvieron tasas de respuesta comparables a las del antígeno de recuerdo del tétanos (39 % frente al 42 %), tasas reducidas en comparación con la vacuna antineumocócica polisacárida (43 % frente al 82 % hasta al menos 2 serotipos de anticuerpos neumocócicos) y en el neoantígeno de KLH (47 % frente al 93 %) cuando se administraron 6 meses después de rituximab en comparación con pacientes que solo recibían metotrexato. Si durante el tratamiento con rituximab se requiriesen vacunaciones con vacunas inactivadas, estas se deben completar al menos 4 semanas antes de comenzar el próximo ciclo de rituximab.
En la experiencia general del tratamiento repetido con rituximab durante un año en la artritis reumatoide, las proporciones de pacientes con títulos de anticuerpos positivos frente S. pneumoniae, gripe, paperas, rubéola, varicela y toxoide tetánico fueron generalmente similares a las proporciones al inicio del estudio.
Uso concomitante/secuencial de otros FARME en la artritis reumatoide
No se recomienda el uso concomitante de Ruxience y tratamientos antirreumáticos distintos de los especificados en la indicación y posología de la artritis reumatoide.
Se dispone de datos limitados de los ensayos clínicos para evaluar completamente la seguridad del uso secuencial de otros FARME (incluidos los inhibidores del TNF y otros medicamentos biológicos) después del tratamiento con rituximab (ver sección 4.5). Los datos disponibles indican que la tasa de infección clínicamente relevante no cambia cuando tales tratamientos se usan en pacientes previamente tratados con rituximab; sin embargo, se debe vigilar estrechamente a los pacientes para detectar signos de infección si se usan medicamentos biológicos y/o FARME después del tratamiento con rituximab.
Neoplasias malignas
Los medicamentos inmunomoduladores pueden aumentar el riesgo de cáncer. Sin embargo, los datos disponibles no sugieren un mayor riesgo de neoplasia maligna para rituximab utilizado en indicaciones autoinmunes más allá del riesgo de neoplasia maligna ya asociado con la enfermedad autoinmune subyacente.
Excipientes
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente “exento de sodio”.
4.5 - Interacciones con otros medicamentos de RUXIENCE 100 mg Concent. para sol. para perfus.
Actualmente, se dispone de datos limitados sobre posibles interacciones farmacológicas con rituximab.
En pacientes con LLC, la administración concomitante de rituximab con fludarabina o ciclofosfamida no pareció tener efectos sobre la farmacocinética de estos medicamentos. Además, no hubo un efecto aparente de la fludarabina y ciclofosfamida sobre la farmacocinética de rituximab.
La coadministración con metotrexato no tuvo ningún efecto sobre la farmacocinética de rituximab en pacientes con artritis reumatoide.
Los pacientes con títulos de anticuerpos humanos anti-murinos (HAMA) o de anticuerpos antifármaco (ADA) pueden sufrir reacciones alérgicas o de hipersensibilidad cuando se tratan con otros anticuerpos monoclonales de diagnóstico o de tratamiento.
En pacientes con artritis reumatoide, 283 pacientes recibieron tratamiento posterior con un FARME biológico después de rituximab. Mientras tomaban rituximab, la tasa de infección clínicamente relevante en estos pacientes fue de 6,01 por 100 pacientes-año en comparación con 4,97 por 100 pacientes-año después del tratamiento con el FARME biológico.
4.6 - Embarazo y Lactancia de RUXIENCE 100 mg Concent. para sol. para perfus.
Anticoncepción en hombres y mujeres
Debido al largo tiempo de permanencia de rituximab en el organismo en pacientes con disminución de células B, las mujeres en edad fértil deben usar métodos anticonceptivos efectivos durante el tratamiento con Ruxience y durante los 12 meses posteriores.
Embarazo
Se sabe que las inmunoglobulinas IgG atraviesan la barrera placentaria.
Los niveles de células B en neonatos humanos después de la exposición materna a rituximab no se han estudiado en los ensayos clínicos. No existen datos adecuados y bien controlados de los estudios en mujeres embarazadas; sin embargo, se han notificado casos de disminución transitoria de células B y linfocitopenia en algunos bebés nacidos de madres expuestas a rituximab durante el embarazo. Se han observado efectos similares en estudios con animales (ver sección 5.3). Por estas razones, Ruxience no se debe administrar a mujeres embarazadas a menos que el posible beneficio esperado supere el riesgo potencial.
Lactancia
Los datos limitados sobre la excreción de rituximab en la leche materna sugieren concentraciones de rituximab en la leche muy bajas (dosis infantil relativa inferior al 0,4 %). Los pocos casos de seguimiento de lactantes describen un crecimiento y desarrollo normal hasta los 2 años. Sin embargo, como estos datos son limitados y los resultados a largo plazo en lactantes se desconocen , no se recomienda la lactancia materna durante el tratamiento con rituximab ni idealmente durante los 6 meses posteriores al tratamiento con rituximab.
Fertilidad
Los estudios en animales no revelaron efectos perjudiciales de rituximab en los órganos reproductores.
4.7 - Efectos sobre la capacidad de conducción de RUXIENCE 100 mg Concent. para sol. para perfus.
No se han realizado estudios sobre los efectos de rituximab sobre la capacidad para conducir y utilizar máquinas, aunque la actividad farmacológica y las reacciones adversas notificadas hasta la fecha indican que la influencia de rituximab sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 - Reacciones Adversas de RUXIENCE 100 mg Concent. para sol. para perfus.
Experiencia en adultos en linfoma no Hodgkin y leucemia linfocítica crónica
Resumen del perfil de seguridad
El perfil de seguridad general de rituximab en el linfoma no Hodgkin y la LLC se basa en los datos de pacientes de ensayos clínicos y los estudios post-comercialización. Estos pacientes fueron tratados con rituximab en monoterapia (como tratamiento de inducción o tratamiento de mantenimiento después del tratamiento de inducción) o en combinación con quimioterapia.
Las reacciones adversas (RAM) observadas con mayor frecuencia en pacientes que recibieron rituximab fueron las RRP, que aparecieron en la mayoría de los pacientes durante la primera perfusión. La incidencia de los síntomas relacionados con la perfusión disminuye sustancialmente con las perfusiones posteriores y es inferior al 1 % después de ocho dosis de rituximab.
Se produjeron acontecimientos infecciosos (predominantemente bacterianos y virales) en aproximadamente el 30-55 % de los pacientes durante los ensayos clínicos en pacientes con LNH y en el 30-50 % de los pacientes durante los ensayos clínicos en pacientes con LLC.
Las reacciones adversas graves notificadas u observadas con mayor frecuencia fueron:
- RRP (incluido el síndrome de liberación de citoquinas y el síndrome de lisis tumoral), ver sección 4.4.
- Infecciones, ver sección 4.4.
- Acontecimientos cardiovasculares, ver sección 4.4.
Otras RAM graves notificadas incluyen la reactivación de la hepatitis B y la LMP (ver sección 4.4.).
Tabla de reacciones adversas
Las frecuencias de las RAM notificadas con rituximab tanto en monoterapia como en combinación con quimioterapia se resumen en la Tabla 3. Las frecuencias se definen como muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras (< 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Las RAM identificadas únicamente durante los estudios post-comercialización, y para las cuales no se pudo estimar una frecuencia, se enumeran como “frecuencia no conocida”.
Tabla 3 RAMnotificadas en ensayos clínicos o durante los estudios post-comercialización en pacientes con NHL y LLC tratados con rituximab en monoterapia/mantenimiento o en combinación con quimioterapia
| Clasificación por órganos y sistemas de MedDRA | Muy frecuentes | Frecuentes | Poco frecuentes | Raras | Muy raras | Frecuencia no conocida |
| Infecciones e infestaciones | infecciones bacterianas, infecciones víricas, +bronquitis | sepsis, +neumonía, +infección febril, +herpes zóster, +infección respiratoria, infección fúngica, infecciones de etiología desconocida,+bronquitis aguda, +sinusitis, hepatitis B1 | infección viral grave2, Pneumocystis jirovecii | LMP | meningoencefalitis enteroviral2, 3 | |
| Trastornos de la sangre y del sistema linfático | neutropenia, leucopenia, +neutropenia febril, trombocitopenia | anemia, +pancitopenia, granulocitopenia | trastornos de la coagulación, anemia aplásica, anemia hemolítica, linfadenopatía | aumento transitorio de los niveles séricos de IgM4 | neutropenia tardía4 | |
| Trastornos del sistema inmunológico | reacciones relacionadas con la perfusión5, angioedema | hipersensibilidad | anafilaxia | síndrome de lisis tumoral, síndrome de liberación de citoquinas5, enfermedad del suero | trombocitopenia aguda reversible relacionada con la perfusión5 | |
| Trastornos del metabolismo y de la nutrición | hiperglucemia, disminución de peso, edema periférico, edema facial, aumento de la LDH, hipocalcemia | |||||
| Trastornos psiquiátricos | depresión, nerviosismo | |||||
| Trastornos del sistema nervioso | parestesia, hipoestesia, agitación, insomnio, vasodilatación, mareos, ansiedad | disgeusia | neuropatía periférica, parálisis facial6 | neuropatía craneal, pérdida de otros sentidos6 | ||
| Trastornos oculares | trastorno de lagrimeo, conjuntivitis | pérdida de visión grave6 | ||||
| Trastornos del oído y del laberinto | acúfenos, dolor de oído | pérdida de audición6 | ||||
| Trastornos cardíacos | +infarto de miocardio5, 7, arritmia, +fibrilación auricular, taquicardia, +trastorno cardíaco | insuficiencia ventricular izquierda, +taquicardia supraventricular, +taquicardia ventricular, +angina, +isquemia miocárdica, bradicardia | trastornos cardíacos graves 5, 7 | insuficiencia cardíaca5, 7 | ||
| Trastornos vasculares | hipertensión arterial, hipotensión ortostática, hipotensión arterial | vasculitis (predominante mente cutánea), vasculitis leucocitoclástica | ||||
| Trastornos respiratorios, torácicos y mediastínicos | Broncoespasmo5, enfermedad respiratoria, dolor torácico, disnea, aumento de tos, rinitis | asma, bronquiolitis obliterante, trastorno pulmonar, hipoxia | enfermedad pulmonar intersticial8 | insuficiencia respiratoria5 | infiltración pulmonar | |
| Trastornos gastrointestinales | náuseas | vómitos, diarrea, dolor en el abdomen, disfagia, estomatitis, estreñimiento, dispepsia, anorexia, irritación de garganta | agrandamiento abdominal | perforación gastrointestinal8 | ||
| Trastornos de la piel y del tejido subcutáneo | prurito, erupción, +alopecia | urticaria, sudoración, sudores nocturnos, +trastorno de la piel | reacciones cutáneas ampollosas graves, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica (síndrome de Lyell)8 | |||
| Trastornos musculoesqueléticos y del tejido conjuntivo | hipertonía, mialgia, artralgia, dolor de espalda, dolor de cuello, dolor | |||||
| Trastornos renales y urinarios | insuficiencia renal5 | |||||
| Trastornos generales y alteraciones en el lugar de administración | fiebre, escalofríos, astenia, dolor de cabeza | dolor tumoral, rubor, malestar general, síndrome del resfriado, +cansancio, +temblores, +insuficiencia multiorgánica5 | dolor en la zona de perfusión | |||
| Exploraciones complementarias | disminución de los niveles de IgG | |||||
| El cálculo de la frecuencia de cada término se basó en todos los grados de la reacción (de leve a grave), a excepción de los términos marcados con “+” donde el cálculo de la frecuencia se basó únicamente en las reacciones graves (≥ grado 3 de los criterios comunes de toxicidad del Instituto Nacional del Cáncer de los EE. UU [NCI]). Únicamente se presenta la frecuencia más alta observada en los ensayos. 1. Incluye reactivación y primoinfecciones; frecuencia basada en el régimen de R-FC en la LLC recidivante/refractaria. 2. Ver también la sección “Infecciones” a continuación. 3. Observada durante la vigilancia postcomercialización. 4. Ver también la sección “Reacciones adversas hematológicas” a continuación. 5. Ver también la sección “Reacciones relacionadas con la perfusión” a continuación. Raramente se han notificado casos mortales. 6. Signos y síntomas de neuropatía craneal. Aparecieron en varios momentos hasta varios meses después de la finalización del tratamiento con rituximab. 7. Observados principalmente en pacientes con afección cardíaca previa y/o quimioterapia cardiotóxica y se relacionaron principalmente con reacciones relacionadas con la perfusión. 8. Incluye casos mortales. | ||||||
Los siguientes términos se notificaron como acontecimientos adversos durante los ensayos clínicos; sin embargo, se notificaron con una incidencia similar o menor en los brazos de rituximab en comparación con los brazos control: hematotoxicidad, infección neutropénica, infección en el tracto urinario, trastorno sensorial, fiebre.
Se notificaron signos y síntomas indicativos de una reacción relacionada con la perfusión en más del 50 % de los pacientes en los ensayos clínicos, y se observaron predominantemente durante la primera perfusión, generalmente en las primeras una o dos horas. Estos síntomas incluían principalmente fiebre, escalofríos y rigidez. Otros síntomas incluyen rubor, angioedema, broncoespasmo, vómitos, náuseas, urticaria/rash, fatiga, cefalea, irritación de garganta, rinitis, prurito, dolor, taquicardia, hipertensión, hipotensión, disnea, dispepsia, astenia y características del síndrome de lisis tumoral. Las reacciones graves relacionadas con la perfusión (como broncoespasmo, hipotensión arterial) ocurrieron hasta en hasta el 12 % de los casos.
Otras reacciones notificadas en algunos casos fueron infarto de miocardio, fibrilación auricular, edema pulmonar y trombocitopenia aguda reversible. Se notificaron con menor frecuencia o frecuencia desconocida el empeoramiento de las afecciones cardíacas preexistentes como angina de pecho o insuficiencia cardíaca congestiva o trastornos cardíacos graves (insuficiencia cardíaca, infarto de miocardio, fibrilación auricular), edema pulmonar, insuficiencia multiorgánica, síndrome de lisis tumoral, síndrome de liberación de citoquinas, insuficiencia renal e insuficiencia respiratoria. La incidencia de los síntomas relacionados con la perfusión disminuyó sustancialmente con las perfusiones posteriores y afectaron a < 1 % de los pacientes en el octavo ciclo del tratamiento con rituximab.
Descripción de reacciones adversas seleccionadas
Infecciones
Rituximab induce la disminución de los células B en el 70-80 % de los pacientes, pero se asoció con una disminución de las inmunoglobulinas séricas solo en una minoría de pacientes.
En el brazo de rituximab de los estudios aleatorizados, se notificaron con una mayor incidencia las infecciones por cándida localizadas, así como el herpes zóster. . Se notificaron infecciones graves en aproximadamente el 4 % de los pacientes tratados con rituximab en monoterapia. Se observaron frecuencias más altas de infecciones globales, incluidas las infecciones de grado 3 o 4, durante el tratamiento de mantenimiento de hasta 2 años con rituximab en comparación con el brazo de observación. No hubo toxicidad acumulativa en términos de infecciones notificadas durante un período de tratamiento de 2 años. Además, en los pacientes tratados con rituximab, se han notificado otras infecciones virales graves, ya sean nuevas, reactivadas o exacerbadas, algunas de las cuales fueron mortales. La mayoría de los pacientes habían recibido rituximab en combinación con quimioterapia o como parte de un trasplante de células madre hematopoyéticas. Ejemplos de estas infecciones virales graves son las infecciones causadas por los virus del herpes (Citomegalovirus, virus de la Varicela Zóster y virus del Herpes Simple), virus JC (leucoencefalopatía multifocal progresiva [LMP]), enterovirus (meningoencefalitis) y virus de la hepatitis C (ver sección 4.4). En los ensayos clínicos también se han notificado casos mortales de LMP tras progresión de la enfermedad y retratamiento. Se han notificado casos de reactivación de la hepatitis B, la mayoría de los cuales fueron en pacientes que recibieron rituximab en combinación con quimioterapia citotóxica. En pacientes con LLC recidivante/refractaria, la incidencia de infección por hepatitis B de grado 3/4 (reactivación y primoinfección) fue del 2 % en R-FC frente al 0 % FC. Se ha observado progresión del sarcoma de Kaposi en pacientes expuestos a rituximab con sarcoma de Kaposi preexistente. Estos casos aparecieron en indicaciones no aprobadas y la mayoría de los pacientes eran VIH positivos.
Reacciones adversas hematológicas
En los ensayos clínicos con rituximab en monoterapia administrado durante 4 semanas, las anomalías hematológicas que aparecieron en una minoría de pacientes fueron, en general, leves y reversibles. Se notificó neutropenia grave (grado 3/4) en el 4,2 %, anemia en el 1,1 % y trombocitopenia en el 1,7 % de los pacientes. Durante el tratamiento de mantenimiento con rituximab de hasta 2 años, se notificaron con una mayor incidencia leucopenia grado 3/4, (5 % vs 2 %,) y neutropenia grado 3/4 (10 % vs 4 %) en comparación con el brazo de observación. La incidencia de trombocitopenia fue baja (< 1 %, grado 3/4) y no hubo diferencias entre los brazos de tratamiento. Durante el ciclo del tratamiento en estudios con rituximab en combinación con quimioterapia, normalmente se notificó con una mayor incidencia de leucopenia de grado 3/4 (R-CHOP 88 % vs CHOP 79 %, R-FC 23 % vs FC 12 %), neutropenia (R-CVP 24 % vs CVP 14 %; R-CHOP 97 % vs CHOP 88 %, R-FC 30 % vs FC 19 % en LLC no tratada previamente), pancitopenia (R-FC 3 % vs FC 1 % en LLC previamente no tratada) en comparación con la quimioterapia sola. Sin embargo, la mayor incidencia de neutropenia en pacientes tratados con rituximab y quimioterapia no se asoció con una mayor incidencia de infecciones e infestaciones en comparación con los pacientes tratados con quimioterapia sola. Los estudios en LLC recidivante/refractaria y no tratada previamente han establecido que hasta en un 25 % de los pacientes tratados con R-FC se prolongó la neutropenia (definida como un recuento de neutrófilos que permaneció por debajo de 1 x 109/l entre los días 24 y 42 después de la última dosis) o fue de aparición tardía (definida como un recuento de neutrófilos por debajo de 1 x 109/l después de 42 días tras la última dosis en pacientes sin neutropenia prolongada previa o que se recuperaron antes del día 42) tras el tratamiento con rituximab en combinación con FC. No se notificaron diferencias para la incidencia de la anemia. Se notificaron algunos casos de neutropenia tardía que aparecieron más de cuatro semanas después de la última perfusión de rituximab. En el estudio de primera línea de LLC, los pacientes con estadio C de Binet experimentaron más acontecimientos adversos en el grupo R-FC en comparación con el grupo FC (R-FC 83 % vs FC 71 %). En el estudio de LLC recidivante/refractaria, se notificó trombocitopenia de grado 3/4 en el 11 % de los pacientes en el grupo de R-FC en comparación con el 9 % de los pacientes en el grupo de FC.
En estudios de rituximab en pacientes con macroglobulinemia de Waldenström, se observaron aumentos transitorios en los niveles séricos de IgM después del inicio del tratamiento, que pueden estar asociados con hiperviscosidad y síntomas relacionados. El aumento transitorio de IgM generalmente descendió hasta al menos el nivel basal en 4 meses.
Reacciones adversas cardiovasculares
Durante los ensayos clínicos con rituximab en monoterapia. se notificaron reacciones cardiovasculares en el 18,8 % de los pacientes, siendo la hipotensión e hipertensión, las reacciones más frecuentes. Se notificaron casos de arritmia de grado 3 o 4 (incluida taquicardia ventricular y supraventricular) y de angina de pecho durante la perfusión. Durante el tratamiento de mantenimiento, la frecuencia de trastornos cardíacos de grado 3/4 fue comparable entre los pacientes tratados con rituximab y los del brazo de observación. Los acontecimientos cardíacos se notificaron como acontecimientos adversos graves (como fibrilación auricular, infarto de miocardio, insuficiencia ventricular izquierda, isquemia miocárdica) en el 3 % de los pacientes tratados con rituximab en comparación con < 1 % en el brazo de control. En los estudios que evaluaron rituximab en combinación con quimioterapia, la incidencia de arritmias cardíacas de grado 3 y 4, predominantemente arritmias supraventriculares como taquicardia y aleteo/fibrilación auricular, fue mayor en el grupo R-CHOP (14 pacientes, 6,9 %) en comparación con el grupo CHOP (3 pacientes, 1,5 %). Todas estas arritmias ocurrieron en el contexto de una perfusión de rituximab o se relacionaron con condiciones predisponentes como fiebre, infección, infarto agudo de miocardio o enfermedad respiratoria y cardiovascular preexistente. No se observaron diferencias entre el grupo R-CHOP y el grupo CHOP en la incidencia de otros acontecimientos cardíacos de grado 3 y 4, incluyendo la insuficiencia cardíaca, la enfermedad miocárdica y manifestaciones de enfermedad coronaria. En la LLC, la incidencia global de los trastornos cardíacos de grado 3 o 4 fue menor tanto en el estudio de primera línea (4 % R-FC, 3 % FC) como en el estudio con LLC recidivante/refractaria (4 % R-FC, 4 % FC).
Sistema respiratorio
Se han notificado casos de enfermedad pulmonar intersticial, algunos con resultado de muerte.
Trastornos neurológicos
Durante el período de tratamiento (fase de tratamiento de inducción que incluye R-CHOP durante un máximo de ocho ciclos), cuatro pacientes (2 %) tratados con R-CHOP, todos con factores de riesgo cardiovascular, sufrieron accidentes cerebrovasculares tromboembólicos durante el primer ciclo de tratamiento. No hubo diferencias en la incidencia de acontecimientos tromboembólicos entre los grupos de tratamiento . Por el contrario, tres pacientes (1,5 %) tuvieron acontecimientos cerebrovasculares en el grupo de CHOP, todos ellos aparecieron durante el período de seguimiento. En la LLC, la frecuencia general de trastornos del sistema nervioso de grado 3 o 4 fue menor tanto en el estudio de primera línea (4 % R-FC, 4 % FC) como en el estudio de LLC recidivante/refractaria (3 % R-FC, 3 % FC).
Se han notificado casos de síndrome de encefalopatía posterior reversible (SEPR)/síndrome de leucoencefalopatía posterior reversible (SLPR). Los signos y síntomas incluyeron alteraciones en la visión, cefalea, convulsiones y alteración del estado mental, con o sin hipertensión relacionada. El diagnóstico de SEPR/SLPR debe confirmarse, mediante técnicas de imagen cerebral.. En los casos notificados, se han reconocido factores de riesgo para SEPR/SLPR, incluyendo la enfermedad subyacente de los pacientes, hipertensión, terapia inmunosupresora y/o quimioterapia.
Trastornos gastrointestinales
Se ha observado perforación gastrointestinal, que en algunos casos fue mortal, en pacientes que tratados con rituximab para el linfoma no Hodgkin. En la mayoría de estos casos, rituximab se administró en combinación con quimioterapia.
Niveles de IgG
En el ensayo clínico que evaluaba el tratamiento de mantenimiento con rituximab en el linfoma folicular recidivante/refractario, la mediana de los niveles de IgG estaba por debajo del límite inferior de la normalidad (LIN) (< 7 g/l) después del tratamiento de inducción tanto en el grupo de observación como en el grupo de rituximab. En el grupo de observación, la mediana del nivel de IgG aumentó posteriormente por encima del LIN, pero permaneció constante en el grupo de rituximab. La proporción de pacientes con niveles de IgG por debajo del LIN fue de aproximadamente el 60 % en el grupo de rituximab durante los 2 años de tratamiento, mientras que disminuyó en el grupo de observación (36 % después de 2 años).
En pacientes pediátricos tratados con rituximab, se ha observado un pequeño número de casos espontáneos y en la literatura médica de hipogammaglobulinemia, en algunos casos grave, y que requiere terapia de reemplazo de inmunoglobulinas a largo plazo. Se desconocen las consecuencias de la depleción prolongada de los células B en pacientes pediátricos.
Trastornos de la piel y del tejido subcutáneo
Se han notificado muy raramente casos de necrólisis epidérmica tóxica (síndrome de Lyell) y de síndrome de Stevens-Johnson, algunos con desenlace mortal.
Subpoblaciones de pacientes - rituximab en monoterapia
Pacientes de edad avanzada (≥ 65 años)
La incidencia de RAMs de todos los grados y RAMs de grado 3/4 fueron similares en pacientes de edad avanzada en comparación con pacientes más jóvenes (< 65 años).
Enfermedad voluminosa o Bulky
Hubo una mayor incidencia de RAMs de grado 3/4 en pacientes con enfermedad de Bulky que en pacientes sin enfermedad de Bulky (25,6 % vs 15,4 %). La incidencia de RAMs de cualquier grado fue similar en estos dos grupos.
Retratamiento
El porcentaje de pacientes que notificaron RAM en el momento del retratamiento con ciclos posteriores de rituximab fue similar al porcentaje de pacientes que notificaron RAM para el tratamiento inicial (RAM de cualquier grado y grado 3/4).
Subpoblaciones de pacientes – rituximab en terapia de combinación
Pacientes de edad avanzada (≥ 65 años)
La frecuencia de acontecimientos adversos sanguíneos y linfáticos de grado 3/4 fue mayor en pacientes de edad avanzada en comparación con pacientes más jóvenes (< 65 años) con LLC no tratada o recidivante/refractaria.
Experiencia de LBDCG / LB / LLA-B madura / LBL pediátrica
Resumen del perfil de seguridad
Se realizó un estudio aleatorizado multicéntrico abierto con quimioterapia LMB con o sin rituximab en pacientes pediátricos (edad ≥ 6 meses a < 18 años) con LBDCG CD20 positivo/LB/LLA-B madura/LBL en estadio avanzado no tratados previamente.
Un total de 309 pacientes pediátricos recibieron rituximab y fueron incluidos en la población de análisis de seguridad. Los pacientes pediátricos asignados aleatoriamente al brazo de quimioterapia LMB con rituximab, o incluidos en la parte del estudio de brazo único, recibieron rituximab en una dosis de 375 mg/m2 de superficie corporal y recibieron un total de seis perfusiones IV de rituximab (dos durante cada uno de los dos ciclos de inducción y una durante cada uno de los dos ciclos de consolidación del esquema Lymphome Malin B (LMB).
El perfil de seguridad de rituximab en pacientes pediátricos (edad ≥ 6 meses a < 18 años) con LBDCG CD20 positivo/LB/LLA-B madura/LBL en estadio avanzado no tratados previamente fue generalmente consistente en tipo, naturaleza y gravedad con el perfil de seguridad conocido en pacientes adultos con LNH y LLC. La adición de rituximab a quimioterapia supuso mayor riesgo de algunos eventos, incluidas infecciones (incluida la sepsis) en comparación con solo quimioterapia.
Experiencia en artritis reumatoide
Resumen del perfil de seguridad
El perfil de seguridad general de rituximab en la artritis reumatoide se basa en los datos de pacientes de los ensayos clínicos y los estudios post comercialización.
El perfil de seguridad de rituximab en pacientes con artritis reumatoide (AR) de moderada a grave se resume en las siguientes secciones. En los ensayos clínicos, más de 3.100 pacientes recibieron al menos un ciclo de tratamiento con un período de seguimiento de 6 meses a más de 5 años; aproximadamente 2.400 pacientes recibieron dos o más ciclos de tratamiento y más de 1.000 recibieron 5 o más ciclos. La información de seguridad recopilada durante la experiencia post comercialización refleja el perfil esperado de reacciones adversas de los ensayos clínicos para rituximab (ver sección 4.4).
Los pacientes recibieron 2 dosis de 1.000 mg de rituximab separadas por un intervalo de dos semanas; además de metotrexato (10-25 mg/semana). Las perfusiones de rituximab se administraron después de una perfusión intravenosa de 100 mg de metilprednisolona; los pacientes también recibieron tratamiento con prednisona oral durante 15 días.
Tabla de reacciones adversas
Las reacciones adversas se enumeran en la Tabla 4. Las frecuencias se definen como muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras (< 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Las reacciones adversas más frecuentes que se consideraron atribuibles a rituximab consistieron en RRP. La incidencia global de las RRP en los ensayos clínicos fue del 23 % con la primera perfusión y disminuyó en las sucesivas perfusiones. Las RRP graves fueron poco frecuentes (0,5 % de los pacientes) y, en su mayoría, en el ciclo inicial. Además de las reacciones adversas observadas en los ensayos clínicos de AR para rituximab, durante la experiencia post comercialización, se han notificado casos de leucoencefalopatía multifocal progresiva (LMP) (ver sección 4.4) y una reacción similar a la enfermedad del suero.
Tabla 4 Resumen de la reacciones adversas notificadas en los ensayos clínicos o durante los estudios post-comercialización que aparecieron en pacientes con artritis reumatoide que recibieron rituximab
| Clasificación por órganos y sistemas de MedDRA | Muy frecuentes | Frecuentes | Poco frecuentes | Raras | Muy raras | Frecuencia no conocida |
| Infecciones e infestaciones | infección del tracto respiratorio superior, infección del tracto urinario | bronquitis, sinusitis, gastroenteritis, tiña del pie | LMP, reactivación de la hepatitis B | infección viral grave1 meningoenc efalitis enteroviral2 | ||
| Trastornos de la sangre y del sistema linfático | neutropenia3 | neutropenia tardía4 | reacción similar a la enfermedad del suero | |||
| Trastornos del sistema inmunológico | 5reacciones relacionadas con la perfusión (hipertensión arterial, náuseas, erupción cutánea, pirexia, prurito, urticaria, irritación de garganta, sofocos, hipotensión arterial, rinitis, rigidez, taquicardia, cansancio, dolor orofaríngeo, edema periférico, eritema) | 5reacciones relacionadas con la perfusión (edema generalizado, broncoespasmo, sibilancias, edema laríngeo, edema angioneurótico, prurito generalizado, anafilaxia, reacción anafilactoide) | ||||
| Trastornos generales y alteraciones en el lugar de administración | ||||||
| Trastornos del metabolismo y de la nutrición | hipercolester olemia | |||||
| Trastornos psiquiátricos | depresión, ansiedad | |||||
| Trastornos del sistema nervioso | cefalea | parestesia, migraña, mareos, ciática | ||||
| Trastornos cardiacos | angina de pecho, fibrilación auricular, insuficiencia cardíaca, infarto de miocardio | aleteo auricular | ||||
| Trastornos gastrointestinales | dispepsia, diarrea, reflujo gastroesofágico, úlceras en la boca, dolor en la parte superior del abdomen | |||||
| Trastornos de la piel y del tejido subcutáneo | alopecia | necrólisis epidérmica tóxica (síndrome de Lyell), síndrome de Stevens-Johnson7 | ||||
| Trastornos musculoesqueléticos y del tejido conjuntivo | artralgia/dolor musculoesquelético, osteoartritis, bursitis | |||||
| Exploraciones complementarias | niveles de IgM disminuidos6 | niveles de IgG disminuidos6 | ||||
| 1. A continuación, consulte tambien la sección Infecciones 2. Observada durante la vigilancia postcomercialización. 3. La categoría de frecuencia se ha obtenido de los valores de laboratorio recopilados como parte de la monitorización rutinaria de laboratorio en los ensayos clínicos. 4. La categoría de frecuencia se ha obtenido de los datos post-comercialización. 5. Reacciones que aparecen durante o en las 24 horas siguientes a la perfusión. Ver también las “Reacciones relacionadas con la perfusión” a continuación. Las RRP pueden aparecer como resultado de la hipersensibilidad y/o por el mecanismo de acción. 6. Incluye observaciones recopiladas como parte de la monitorización rutinaria de laboratorio. 7. Incluye casos mortales. | ||||||
Ciclos múltiples
Ciclos múltiples de tratamiento muestran un perfil de RAM similar al observado después de la primera exposición. La incidencia de todas las RAM después de la primera exposición a rituximab fue más alta durante los primeros 6 meses y disminuyó posteriormente. Esto se explica principalmente por las RRP (más frecuentes durante el primer ciclo de tratamiento), la exacerbación de la AR y las infecciones, que fueron más frecuentes en los primeros 6 meses de tratamiento.
Descripción de reacciones adversas seleccionadas
Reacciones relacionadas con la perfusión
Las RAM más frecuentes tras la administración de rituximab en los estudios clínicos fueron RRP (consultar la Tabla 4). Entre los 3.189 pacientes tratados con rituximab, 1.135 (36 %) experimentaron al menos una RRP y 733/3.189 (23 %) de los pacientes experimentaron una RRP después de la primera perfusión de la primera exposición a rituximab. La frecuencia de las RRP disminuyó con las sucesivas perfusiones. En los ensayos clínicos, menos del 1 % (17/3.189) de los pacientes experimentaron una RRP grave. No hubo criterios comunes de toxicidad (CCT) grado 4 de RRP ni muertes por RRP en los ensayos clínicos. La proporción de acontecimientos de grado 3 de los CCT y de RRP que condujeron a la retirada del fármaco, disminuyó con cada ciclo y fueron raros a partir del ciclo 3. La premedicación con glucocorticoides intravenosos redujo significativamente la frecuencia y la gravedad de las RRP (ver las secciones 4.2 y 4.4). Durante la comercialización, se han notificado RRP graves con desenlace mortal.
En un ensayo diseñado para evaluar la seguridad de una perfusión de rituximab más rápida en pacientes con artritis reumatoide, a los pacientes con AR activa de moderada a grave que no experimentaron una RRP grave durante o en las 24 horas siguientes a la primera perfusión estudiada se les permitió recibir una perfusión intravenosa de 2 horas de rituximab. Los pacientes con antecedentes de una reacción grave a la perfusión a una terapia biológica para la AR fueron excluidos de la entrar en el ensayo. La incidencia, los tipos y la gravedad de las RRP concordaron con lo observado históricamente. No se observaron RRP graves.
Infecciones
La tasa general de infecciones notificadas en los ensayos clínicos se aproximó al 94 por 100 pacientes-año en los pacientes tratados con rituximab. Las infecciones fueron predominantemente leves a moderadas y afectaron mayoritariamente al tracto respiratorio superior y al tracto urinario. La frecuencia de infecciones que fueron graves o requirieron antibióticos por vía intravenosa fue de aproximadamente 4 por 100 pacientes-año. La incidencia de infecciones graves no mostró ningún aumento significativo después de múltiples ciclos de rituximab. Las infecciones del tracto respiratorio inferior (incluida la neumonía) se han notificado durante los ensayos clínicos, con una incidencia similar en el brazo de rituximab en comparación con el brazo control.
En el entorno posterior a la comercialización, se notificaron infecciones virales graves en pacientes con AR tratados con rituximab.
Se han notificado casos de leucoencefalopatía multifocal progresiva con desenlace mortal tras el uso de rituximab para el tratamiento de enfermedades autoinmunes. Esto incluye la artritis reumatoide y enfermedades autoinmunes fuera de la indicación de Ruxience, incluido el lupus eritematoso sistémico (LES) y la vasculitis.
En pacientes con linfoma no Hodgkin que reciben rituximab en combinación con quimioterapia citotóxica, se han notificado casos de reactivación de la hepatitis B (ver “Linfoma no Hodgkin”). La reactivación de la infección por hepatitis B también se ha notificado muy raramente en pacientes con artritis reumatoide que reciben rituximab (ver sección 4.4).
Reacciones adversas cardiovasculares
Se notificaron reacciones cardíacas graves a una tasa de 1,3 por 100 pacientes-año en los pacientes tratados con rituximab en comparación con 1,3 por 100 pacientes-año en pacientes tratados con placebo. La proporción de pacientes que experimentaron reacciones cardíacas (todas o graves) no aumentó en los ciclos múltiples.
Acontecimientos neurológicos
Se han notificado casos de síndrome de encefalopatía posterior reversible (SEPR)/síndrome de leucoencefalopatía posterior reversible (SLPR). Los signos y síntomas incluyeron trastornos visuales, cefalea, convulsiones y estado mental alterado, con o sin hipertensión relacionada. Un diagnóstico de SEPR/SLPR requiere confirmación mediante escáner cerebral. Los casos notificados tenían factores de riesgo reconocidos para SEPR/SLPR, incluyendo la enfermedad subyacente de los pacientes, hipertensión, terapia inmunosupresora y/o quimioterapia.
Neutropenia
Se observaron acontecimientos de neutropenia con el tratamiento con rituximab, la mayoría de los cuales fueron transitorios y de gravedad leve o moderada. La neutropenia puede ocurrir varios meses después de la administración de rituximab (ver sección 4.4).
En las fases controladas con placebo de los ensayos clínicos, el 0,94 % (13/1.382) de los pacientes tratados con rituximab y el 0,27 % (2/731) de los pacientes tratados con placebo desarrollaron neutropenia grave.
Raramente se notificaron acontecimientos neutropénicos, incluyendo la neutropenia tardía grave y la persistente, en el período post comercialización, algunos de los cuales se relacionaron con infecciones mortales.
Trastornos de la piel y del tejido subcutáneo
Se han notificado muy raramente casos de necrólisis epidérmica tóxica (síndrome de Lyell) y de síndrome de Stevens-Johnson, algunos con desenlace mortal.
Anomalías en las pruebas de laboratorio
En pacientes con AR tratados con rituximab, se ha observado hipogammaglobulinemia (IgG o IgM por debajo del límite inferior de la normalidad). No hubo incremento en la tasa de infecciones global o infecciones graves después de la disminución de IgG o IgM (ver sección 4.4).
En pacientes pediátricos tratados con rituximab, se ha observado un pequeño número de casos espontáneos y en la bibliografía médica, de hipogammaglobulinemia, en algunos casos grave y que requiere terapia prolongada de reposición de inmunoglobulinas. Se desconocen las consecuencias de la depleción prolongada de las células B en pacientes pediátricos.
Experiencia en granulomatosis con poliangeitis (GPA) y poliangeitis microscópica (PAM)
Inducción de la remisión en adultos (Estudio 1 GPA/PAM)
En el estudio 1 GPA/PAM, se trató a 99 pacientes adultos para inducir la remisión de GPA y PAM con rituximab (375 mg/m2, una vez por semana durante 4 semanas) y glucocorticoides (ver sección 5.1).
Las RAM enumeradas en la Tabla 5 fueron todas reacciones adversas que ocurrieron con una incidencia ≥ 5 % y con una frecuencia más alta en el grupo de rituximab que el grupo de comparación.
Tabla 5 Reacciones adversas que aparecen a los 6 meses en ≥ 5 % de los pacientes adultos que recibieron rituximab en el estudio 1 GPA/PAM (Rituximab n=99), con mayor frecuencia que con el grupo comparador, o durante la vigilancia poscomercialización
| Clasificación por órganos y sistemas de MedDRA Reacciones adversas | Frecuencia | |
| Infecciones e infestaciones | ||
| Infecciones urinarias | 7 % | |
| Bronquitis | 5 % | |
| Herpes zóster | 5 % | |
| Nasofaringitis | 5 % | |
| Infección viral grave1,2 | No conocida | |
| Meningoencefalitis enteroviral1 | No conocida | |
| Trastornos de la sangre y del sistema linfático | ||
| Trombocitopenia | 7 % | |
| Trastornos del sistema inmunológico | ||
| Síndrome de liberación de citoquinas | 5 % | |
| Trastornos del metabolismo y de la nutrición | ||
| Hiperpotasemia | 5 % | |
| Trastornos psiquiátricos | ||
| Insomnio | 14 % | |
| Trastornos del sistema nervioso | ||
| Mareos | 10 % | |
| Temblor | 10 % | |
| Trastornos vasculares | ||
| Hipertensión | 12 % | |
| Rubor | 5 % | |
| Trastornos respiratorios, torácicos y mediastínicos | ||
| Tos | 12 % | |
| Disnea | 11 % | |
| Epistaxis | 11 % | |
| Congestión nasal | 6 % | |
| Trastornos gastrointestinales | ||
| Diarrea | 18 % | |
| Dispepsia | 6 % | |
| Estreñimiento | 5 % | |
| Trastornos de la piel y del tejido subcutáneo | ||
| Acné | 7 % | |
| Trastornos musculoesqueléticos y del tejido conjuntivo | ||
| Calambres musculares | 18 % | |
| Artralgia | 15 % | |
| Dolor de espalda | 10 % | |
| Debilidad muscular | 5 % | |
| Dolor musculoesquelético | 5 % | |
| Dolor en las extremidades | 5 % | |
| Trastornos generales y alteraciones en el lugar de administración | ||
| Edema periférico | 16 % | |
| Exploraciones complementarias | ||
| Hemoglobina disminuida | 6 % | |
| 1 Observado durante la vigilancia poscomercialización. 2 Ver también la sección Infecciones más abajo. | ||
Tratamiento de mantenimiento en adultos (Estudio 2 GPA/PAM)
En el estudio 2 GPA/PAM, un total de 57 pacientes adultos con GPA y PAM activas y graves fueron tratados con rituximab para el mantenimiento de la remisión (ver sección 5.1).
Tabla 6 Reacciones adversas que aparecen en ≥ 5 % de los pacientes adultos que reciben rituximab en el estudio 2 GPA/PAM (Rituximab n=57), con mayor frecuencia que el grupo comparador, o durante la vigilancia poscomercialización
| Clasificación por órganos y sistemas de MedDRA Reacciones adversas | Frecuencia | |
| Infecciones e infestaciones | ||
| Bronquitis | 14 % | |
| Rinitis | 5 % | |
| Infección viral grave1,2 | no conocida | |
| Meningoencefalitis enteroviral1 | No conocida | |
| Trastornos respiratorios, torácicos y mediastínicos | ||
| Disnea | 9 % | |
| Trastornos gastrointestinales | ||
| Diarrea | 7 % | |
| Trastornos generales y alteraciones en el lugar de administración | ||
| Pirexia | 9 % | |
| Síndrome pseudogripal | 5 % | |
| Edema periférico | 5 % | |
| Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | ||
| Reacciones relacionadas con la perfusión2 | 12 % | |
| 1Observado durante la vigilancia poscomercialización. 2 Ver también la sección “Infecciones” más abajo. 3 Se proporcionan detalles de las reacciones relacionadas con la perfusión en la sección “Descripción de reacciones adversas seleccionadas”. | ||
El perfil de seguridad general fue acorde con el perfil de seguridad ya establecido para rituximab en las indicaciones autoinmunes aprobadas, incluyendo GPA/PAM. En general, el 4 % de los pacientes en el brazo de rituximab experimentaron acontecimientos adversos que llevaron a la interrupción del tratamiento. La mayoría de los acontecimientos adversos en el grupo de rituximab fueron de intensidad leve o moderada. Ningún paciente en el brazo de rituximab sufrió acontecimientos adversos mortales.
Los acontecimientos notificados con más frecuencia considerados como RAM fueron las reacciones relacionadas con la perfusión y las infecciones.
Estudio observacional a largo plazo (Estudio 3 GPA/PAM)
En un estudio observacional de seguridad a largo plazo, 97 pacientes con GPA/PAM recibieron tratamiento con rituximab (media de 8 perfusiones [rango 1-28]) durante un máximo de 4 años, de acuerdo con la práctica clínica estándar. El perfil de seguridad global fue consistente con el perfil de seguridad ya establecido de rituximab en AR y GPA/PAM y no se comunicaron nuevas reacciones adversas.
Población pediátrica
Se realizó un estudio abierto de un solo brazo en 25 pacientes pediátricos con GPA o con PAM, activas y graves. El período de estudio general consistió en una fase de inducción de remisión de 6 meses con seguimiento de 18 meses como mínimo, hasta 4,5 años en total. Durante la fase de seguimiento, rituximab se administró a discreción del investigador (17 de los 25 pacientes recibieron tratamiento adicional con rituximab). Se permitió el tratamiento concomitante con otra terapia inmunosupresora (ver sección 5.1).
Las RAM fueron consideradas como eventos adversos que ocurrieron con una incidencia de ≥ 10 %. Estos incluyeron: infecciones (17 pacientes [68 %] en la fase de inducción de remisión; 23 pacientes [92 %] en el período de estudio general), RRP (15 pacientes [60 %] en la fase de inducción de remisión; 17 pacientes [68 %] en el período de estudio general) y náuseas (4 pacientes [16 %] en la fase de inducción de remisión; 5 pacientes [20 %] en el período de estudio general).
Durante el período de estudio general, el perfil de seguridad de rituximab fue consistente con el reportado durante la fase de inducción de remisión.
El perfil de seguridad de rituximab en pacientes pediátricos con GPA o PAM fue consistente en tipo, naturaleza y gravedad con el perfil de seguridad conocido en pacientes adultos en las indicaciones autoinmunes aprobadas, incluyendo la GPA o PAM en adultos.
Descripción de reacciones adversas seleccionadas
Reacciones relacionadas con la perfusión
En el estudio 1 de GPA/PAM (estudio de inducción de remisión en adultos), las RRP se definieron como cualquier evento adverso que apareciera dentro de las 24 horas tras la perfusión y que los investigadores las considerasen relacionadas con la perfusión en la población de seguridad. De los 99 pacientes tratados con rituximab, 12 (12 %) experimentaron al menos una RRP. Todas las RRP fueron CCT grado 1 o 2. Las RRP más frecuentes incluyeron síndrome de liberación de citoquinas, rubor, irritación de garganta y temblor. Rituximab se administró en combinación con glucocorticoides por vía intravenosa los cuales pueden reducir la incidencia y la gravedad de estos eventos.
En el estudio 2 de GPA/PAM (estudio de mantenimiento en adultos), 7/57 (12 %) pacientes en el brazo de rituximab experimentaron al menos una reacción relacionada con la perfusión. La incidencia de los síntomas de RRP fue mayor durante o después de la primera perfusión (9 %) y disminuyó con las sucesivas perfusiones (< 4 %). Todos los síntomas de las RRP fueron leves o moderados y la mayoría de ellos se notificaron según la clasificación SOC como “Trastornos respiratorios, torácicos y mediastínicos” y “Trastornos de la piel y del tejido subcutáneo”.
En el ensayo clínico en pacientes pediátricos con GPA o PAM, las RRP informadas se observaron predominantemente con la primera perfusión (8 pacientes [32 %]) y luego disminuyeron con el tiempo con el número de perfusiones de rituximab (20 % con la segunda perfusión, 12 % con la tercera perfusión y 8 % con la cuarta perfusión). Los síntomas de RRP más comunes notificados durante la fase de inducción de remisión fueron: dolor de cabeza, erupción cutánea, rinorrea y pirexia (8%, para cada síntoma). Los síntomas observados de RRP fueron similares a los conocidos en pacientes adultos con GPA o PAM tratados con rituximab. La mayoría de las RRP fueron de Grado 1 y Grado 2, hubo dos RRP de grado 3 no graves y no se informaron RRP de grado 4 o 5. En un paciente se informó una RRP de grado 2 grave (edema generalizado que se resolvió con el tratamiento) (ver sección 4.4).
Infecciones
En el estudio 1 de GPA/PAM, la tasa global de infección fue aproximadamente de 237 por 100 pacientes-año (IC del 95 % 197-285) a los 6 meses en la variable principal . Las infecciones fueron predominantemente de leves a moderadas y afectaron mayoritariamente al tracto respiratorio superior, herpes zóster y al tracto urinario. La incidencia de infecciones graves fue de aproximadamente 25 por 100 pacientes-año. La infección grave más frecuentemente notificada en el grupo de rituximab fue la neumonía con una frecuencia del 4 %.
En el estudio 2 de GPA/PAM, 30/57 (53 %) pacientes en el brazo de rituximab experimentaron infecciones. La incidencia de las infecciones de cualquier grado fue similar entre los brazos. Las infecciones fueron en su mayoría de leve a moderadas. Las infecciones más frecuentes en el brazo de rituximab incluyeron infecciones del tracto respiratorio superior, gastroenteritis, infecciones del tracto urinario y herpes zóster. La incidencia de las infecciones graves fue similar en ambos brazos (aproximadamente el 12 %). La infección grave notificada con más frecuencia en el grupo de rituximab fue bronquitis de leve o moderada.
En el ensayo clínico en pacientes pediátricos con GPA y PAM, activas y graves, el 91 % de las infecciones notificadas no fueron graves y el 90 % fueron de leves a moderadas.
Las infecciones más comunes en la fase general fueron: infecciones del tracto respiratorio superior (ITRS)(48 %), influenza (24 %), conjuntivitis (20 %), nasofaringitis (20 %), infecciones del tracto respiratorio inferior (16 %), sinusitis (16 %), ITRS virales (16 %), infección de oído (12 %), gastroenteritis (12 %), faringitis (12 %), infección del tracto urinario (12 %). Se informaron infecciones graves en 7 pacientes (28 %) e incluyeron: influenza (2 pacientes [8 %]) e infección del tracto respiratorio inferior (2 pacientes [8 %]) como los eventos informados con mayor frecuencia.
En el entorno posterior a la comercialización, se notificaron infecciones virales graves en pacientes con GPA/PAM que habían sido tratados con rituximab.
Tumores
En el estudio 1 de GPA/PAM, la incidencia de tumores en los pacientes con GPA y PAM tratados con rituximab fue de 2,00 por cada 100 pacientes-año en la fecha de cierre común del estudio (cuando el último paciente ha terminado el período de seguimiento). Basándose en las tasas incidencia estandarizados, la incidencia de tumores parece similar a la notificada previamente en pacientes con vasculitis asociada a ANCA.
En el ensayo clínico pediátrico, no se notificaron tumores malignos con un período de seguimiento de hasta 54 meses.
Reacciones adversas cardiovasculares
En el estudio 1 de GPA/MPA, los eventos cardíacos aparecieron con una tasa de aproximadamente 273 por 100 pacientes-año (IC del 95 % 149-470) a los 6 meses en la variable principal. La tasa de eventos cardíacos graves fue de 2,1 por 100 pacientes-año (IC del 95 % 3-15). Los eventos notificados con mayor frecuencia fueron taquicardia (4 %) y fibrilación auricular (3 %) (ver sección 4.4).
Acontecimientos neurológicos
Se han notificado casos de síndrome de encefalopatía posterior reversible (SEPR)/síndrome de leucoencefalopatía posterior reversible (SLPR) en afecciones autoinmunes. Los signos y síntomas incluyeron alteraciones visuales, cefalea, convulsiones y alteración del estado mental, con o sin hipertensión asociada. Un diagnóstico de SEPR/SLPR requiere confirmación mediante escáner cerebral. Los casos notificados de SEPR/SLPR tenían factores de riesgo reconocidos incluyendo la enfermedad subyacente de los pacientes, la hipertensión, y la terapia de inmunosupresión y/o quimioterapia.
Reactivación de la hepatitis B
Se ha notificado un pequeño número de casos de reactivación de la hepatitis B, algunos con resultado de muerte, en pacientes con granulomatosis con poliangeitis y poliangeitis microscópica que reciben rituximab en el período post comercialización.
Hipogammaglobulinemia
Se ha observado hipogammaglobulinemia (IgA, IgG o IgM por debajo del límite inferior normal) en pacientes adultos y pediátricos con GPA y PAM tratados con rituximab.
En el estudio 1 de GPA/PAM, a los 6 meses, el 27 %, 58 % y 51 % de los pacientes del grupo de rituximab con niveles normales de inmunoglobulina al inicio del estudio tenían niveles bajos de IgA, IgG e IgM, respectivamente, en comparación con el 25 %, 50 % y 46 % de los pacientes del grupo de ciclofosfamida. La tasa de infecciones generales e infecciones graves no aumentó después del desarrollo de niveles bajos de IgA, IgG o IgM.
En el estudio 2 de GPA/PAM, no hubo diferencias clínicamente significativas entre los dos brazos de tratamiento ni se observó una disminución en los niveles totales de inmunoglobulinas, IgG, IgM o IgA durante el ensayo.
En el ensayo clínico pediátrico, durante el período de estudio general, 3/25 (12 %) pacientes notificaron un evento de hipogammaglobulinemia, 18 pacientes (72 %) tuvieron niveles bajos de IgG (definidos como niveles de Ig por debajo del límite normal durante al menos 4 meses) prolongados en el tiempo (de los cuales 15 pacientes también tuvieron niveles bajos de IgM prolongados). Tres pacientes recibieron tratamiento con inmunoglobulina intravenosa (IG-IV).
En base a los limitados datos recopilados, no se pueden extraer conclusiones firmes de si niveles bajos prolongados de IgG e IgM conducen a un mayor riesgo de infección grave en estos pacientes. Se desconocen las consecuencias de la depleción de las células B a largo plazo en pacientes pediátricos.
Neutropenia
En el estudio 1 de GPA/PAM, el 24 % de los pacientes en el grupo de rituximab (ciclo único) y el 23 % de los pacientes en el grupo de ciclofosfamida desarrollaron neutropenia de CCT grado 3 o superior. La neutropenia no se asoció con un aumento observado en la infección grave en pacientes tratados con rituximab.
En el estudio 2 de GPA/PAM, la incidencia de neutropenia de cualquier grado fue del 0 % para los pacientes tratados con rituximab frente al 5 % para los pacientes tratados con azatioprina.
Trastornos de la piel y del tejido subcutáneo
Se han notificado muy raramente casos de necrólisis epidérmica tóxica (síndrome de Lyell) y de síndrome de Stevens-Johnson, algunos con desenlace mortal.
Experiencia en pénfigo vulgar
Resumen del perfil de seguridad en el Estudio 1 de PV (Estudio ML22196) y en el Estudio 2 de PV (WA29330)
El perfil de seguridad de rituximab en combinación con glucocorticoides, a dosis baja y corto plazo en el tratamiento de pacientes con pénfigo vulgar, se estudió en un estudio en Fase III, aleatorizado, controlado, multicéntrico y abierto que incluyó a 38 pacientes con pénfigo vulgar (PV) distribuidos aleatoriamente en el grupo de rituximab (Estudio 1 de PV). Los pacientes incluidos en el grupo de rituximab recibieron una dosis inicial de 1.000 mg IV el día 1 del estudio y una segunda dosis intravenosa de 1.000 mg el día 15 del estudio. Se administraron dosis de mantenimiento de 500 mg IV a los 12 y 18 meses. Los pacientes podían recibir 1.000 mg IV en el momento de la recidiva (ver sección 5.1).
En el Estudio 2 de PV, un estudio aleatorizado, doble ciego, doble simulado, de comparación activa y multicéntrico que evalúa la eficacia y seguridad de rituximab en comparación con micofenolato mofetilo (MFM) en pacientes con PV de moderado a grave que requieren corticosteroides orales, 67 pacientes con PV recibieron tratamiento con rituximab (1000 mg IV iniciales en el día 1 del estudio y una segunda dosis de 1000 mg IV en el día 15 del estudio repetidos en las semanas 24 y 26) durante hasta 52 semanas (ver sección 5.1).
El perfil de seguridad de rituximab en pacientes con PV fue consistente con perfil de seguridad establecido en otras indicaciones autoinmunes aprobadas.
Tabla de reacciones adversas para los estudios de PV 1 y 2
Las reacciones adversas de los estudios de PV 1 y 2 presentadas en la Tabla 7. En el estudio 1 de PV, las RAM fueron definidas como acontecimientos adversos que ocurrieron con una tasa de ≥ 5 % entre los pacientes con PV tratados con rituximab, con una diferencia absoluta de ≥ 2 % en la incidencia entre el grupo tratado con rituximab y el grupo de prednisona a dosis estándar hasta el mes 24. Ningún paciente fue retirado del ensayo debido a las RAM en el Estudio 1 de PV. En el Estudio 2 de PV, las RAM corresponden a acontecimientos adversos que ocurrieron en una tasa de ≥ 5 % en el brazo de rituximab y se evaluaron como relacionados.
Tabla 7 Reacciones adversas para pacientes con pénfigo vulgar tratados con rituximab en el estudio 1 de PV (hasta el mes 24) y en el estudio 2 de PV (hasta la semana 52), o durante la vigilancia poscomercialización
| Clasificación por órganos y sistemas de MedDRA | Muy frecuentes | Frecuentes | No frecuentes |
| Infecciones e infestaciones | Infección del tracto respiratorio superior | Infección por virus del herpes Herpes zóster Herpes oral Conjuntivitis Nasofaringitis Candidiasis oral Infección del tracto urinario | Infección viral grave1,2 Meningoencefalitis enteroviral1 |
| Neoplasias benignas, malignas y no especificadas (incl quistes y pólipos) | Papiloma cutáneo | ||
| Trastornos psiquiátricos | Trastornos depresivos persistentes | Depresión mayor Irritabilidad | |
| Trastornos del sistema nervioso | Dolor de cabeza | Mareos | |
| Trastornos cardiacos | Taquicardia | ||
| Trastornos gastrointestinales | Dolor abdominal superior | ||
| Trastornos de la piel y del tejido subcutáneo | Alopecia | Prurito Urticaria Trastornos de la piel | |
| Trastornos musculoesqueléticos y del tejido conjuntivo | Dolor musculoesquelético Artralgia Dolor de espalda | ||
| Trastornos generales y alteraciones en el lugar de administración | Fatiga Astenia Pirexia | ||
| Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | Reacciones relacionadas con la perfusión3 | ||
| 1 Observado durante la vigilancia poscomercialización. 2. Ver también sección infecciones más abajo. 3 Las reacciones relacionadas con la perfusión del estudio 1 de PV incluyeron síntomas recogidos en la siguiente visita programada después de cada perfusión y eventos adversos que ocurrieron el día o un día después de la perfusión. Las reacciones relacionadas con la perfusión/términos comunicados para el estudio 1 de PV más frecuentemente incluyeron síntomas como dolores de cabeza, escalofríos, presión arterial alta, náuseas, astenia y dolor. Las reacciones relacionadas con la perfusión/términos comunicados para el Estudio 2 de PV más frecuentemente incluyeron síntomas como disnea, eritema, hiperhidrosis, rubor/sofocos, hipotensión/presión arterial baja y erupción/erupción cutánea pruriginosa. | |||
Descripción de reacciones adversas seleccionadas
Reacciones relacionadas con la perfusión
Las reacciones relacionadas con la perfusión en el estudio 1 de PV fueron frecuentes (58 %). Casi todas las reacciones relacionadas con la perfusión fueron de leves a moderadas. La proporción de pacientes que experimentaron una reacción relacionada con la perfusión fue del 29 % (11 pacientes), 40 % (15 pacientes), 13 % (5 pacientes) y 10 % (4 pacientes) después de la primera, segunda, tercera y cuarta perfusión, respectivamente. Ningún paciente fue retirado del tratamiento debido a las reacciones relacionadas con la perfusión. Los síntomas de las reacciones relacionadas con la perfusión fueron similares en tipo y gravedad a los observados en pacientes con AR y GPA/PAM.
En el Estudio 2 de PV, las RRP se produjeron principalmente en la primera perfusión y la frecuencia de las RRP disminuyó con las perfusiones posteriores: 17,9 %, 4,5 %, 3 % y 3 % de los pacientes experimentaron RRP en la primera, segunda, tercera y cuarta perfusiones, respectivamente. En 11/15 pacientes que experimentaron al menos una RRP, las RRP fueron de Grado 1 o 2. En 4/15 pacientes, se notificaron RRP de Grado ≥ 3 y supusieron la interrupción del tratamiento con rituximab; tres de los cuatro pacientes experimentaron RRP graves (potencialmente mortales). Se produjeron RRP graves en la primera perfusión (2 pacientes) o segunda (1 paciente) que se resolvieron con tratamiento sintomático.
Infecciones
En el estudio 1 de PV en el grupo de rituximab, 14 pacientes (37 %) experimentaron infecciones relacionadas con el tratamiento en comparación con 15 pacientes (42 %) en el grupo de prednisona a dosis estándar. Las infecciones más frecuentes en el grupo de rituximab fueron las infecciones por herpes simple y zóster, bronquitis, infección del tracto urinario, micosis y conjuntivitis. En el grupo de rituximab, 3 pacientes (8 %) experimentaron un total de 5 infecciones graves (neumonía por Pneumocystis jirovecii, trombosis infecciosa, discitis intervertebral, infección pulmonar, sepsis por Staphylococcus) y un paciente (3 %) en el grupo de dosis estándar de prednisona experimentó una infección grave (neumonía por Pneumocystis jirovecii).
En el estudio 2 de PV, 42 pacientes (62,7 %) en el brazo de rituximab experimentaron infecciones. Las infecciones más comunes en el grupo de pacientes con rituximab fueron infección del tracto respiratorio superior, nasofaringitis, candidiasis oral e infección del tracto urinario. Seis pacientes (9 %) del brazo de rituximab experimentaron infecciones graves.
En el entorno posterior a la comercialización, se notificaron infecciones virales graves en pacientes con PV que habían sido tratados con rituximab.
Anomalías de laboratorio
En el Estudio 2 de PV, en el brazo de rituximab, se observaron muy frecuentemente disminuciones transitorias en el recuento de linfocitos, impulsadas por las disminuciones en las poblaciones periféricas de células T, así como una disminución transitoria en el nivel de fósforo después de la infusión. Estos se consideraron inducidos por perfusión intravenosa de premedicación con metilprednisolona.
En el estudio 2 de PV, se observaron muy frecuentemente niveles bajos de IgG y niveles bajos de IgM; sin embargo, no hubo evidencia de un mayor riesgo de infecciones graves después del desarrollo de baja IgG o IgM.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es
4.9 - Sobredosificación de RUXIENCE 100 mg Concent. para sol. para perfus.
Existe experiencia limitada, en ensayos clínicos en humanos, administrando dosis más altas que la aprobada para la formulación intravenosa de rituximab. La dosis intravenosa más alta de rituximab probada en humanos hasta la fecha es de 5.000 mg (2.250 mg/m2), probada en un estudio con aumento progresivo de la dosis en pacientes con LLC. No se identificaron señales de seguridad adicionales.
A los pacientes que experimentan una sobredosis se les debe interrumpir la perfusión inmediatamente y se les debe monitorizar estrechamente.
Se han notificado cinco casos de sobredosis de rituximab en el período post-comercialización. En tres casos no se notificó ningún acontecimiento adverso. En los otros dos, los acontecimientos adversos notificados fueron síntomas similares a los de la gripe, con una dosis de 1,8 g de rituximab e insuficiencia respiratoria mortal, con una dosis de 2 g de rituximab.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de RUXIENCE 100 mg Concent. para sol. para perfus.
Grupo farmacoterapéutico: agentes antineoplásicos, anticuerpos monoclonales, código ATC: L01FA01
Ruxience es un medicamento biosimilar. La información detallada sobre este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Rituximab se une específicamente al antígeno transmembrana, CD20, una fosfoproteína no glucosilada, situada en los pre-linfocitos B y los linfocitos B maduros. El antígeno se expresa en > 95 % de todos los linfomas no Hodgkin de células B.
CD20 se encuentra tanto en células B normales como en tumorales, pero no en células madre hematopoyéticas, pre-células B, células plasmáticas normales u otro tejido normal. Este antígeno no se internaliza tras la unión del anticuerpo y no se desprende de la superficie celular. CD20 no circula en el plasma como un antígeno libre y, por lo tanto, no compite por la unión del anticuerpo.
El dominio Fab de rituximab se une al antígeno CD20 en los linfocitos B mientras que el dominio Fc puede reclutar efectores de la respuesta inmune para mediar la lisis de las células B. Los posibles mecanismos de la lisis celular mediada por efector incluyen la citotoxicidad dependiente del complemento (CDC) resultante de la unión a C1q y la citotoxicidad celular dependiente de anticuerpos (ADCC) mediada por uno o más receptores Fcγ de la superficie de los granulocitos, macrófagos y células NK. También se ha demostrado que la unión de rituximab al antígeno CD20 en los linfocitos B induce la muerte celular a través de la apoptosis.
Los recuentos de células B periféricos disminuyeron por debajo de lo normal después de completar la primera dosis de rituximab. En los pacientes tratados de neoplasias hematológicas, la recuperación de células B comenzó a los 6 meses de tratamiento y, en general, se recuperaron los niveles normales en los 12 meses después de finalizado el tratamiento, aunque en algunos pacientes puede llevar más tiempo (hasta un tiempo medio de recuperación de 23 meses después de la terapia de inducción). En pacientes con artritis reumatoide, se observó una depleción inmediata de las células B en sangre periférica después de dos perfusiones de 1.000 mg de rituximab separadas por un intervalo de 14 días. Los recuentos de células B en sangre periférica comenzaron a aumentar a partir de la semana 24 y la evidencia de repoblación se observa, en la mayoría de los pacientes, en la semana 40, independientemente de que rituximab se administre en monoterapia o en combinación con metotrexato. Una pequeña proporción de pacientes tuvo una depleción prolongada de células B periféricos que duró 2 años o más tras la última dosis de rituximab. En pacientes con GPA o PAM, el número de linfocitos B de sangre periférica disminuyó a < 10 células/μl después de dos perfusiones semanales de rituximab 375 mg/m2, y se mantuvo en ese nivel en la mayoría de los pacientes a los 6 meses. La mayoría de los pacientes (81 %) mostraron signos de retorno de las células B y recuentos > 10 células/μl a los 12 meses, aumentando al 87 % de los pacientes a los 18 meses.
Experiencia clínica en linfoma no Hodgkin y en leucemia linfocítica crónica
Linfoma folicular
Monoterapia
Tratamiento inicial, semanal, 4 dosis
En el ensayo pivotal, 166 pacientes con LNH recidivante, quimioresistente de bajo grado o folicular de células B recibieron 375 mg/m2 de rituximab como perfusión intravenosa una vez por semana durante cuatro semanas. La tasa de respuesta global (TRG/ORR) en la población por intención de tratar (ITT) fue del 48 % (IC95 %: 41 % - 56 %) con un 6 % de respuestas completas (RC) y un 42 % de respuestas parciales (RP). La mediana proyectada del tiempo hasta progresión (TTP) en los pacientes respondedores fue de 13,0 meses. En un análisis de subgrupos, la TRG fue mayor en pacientes con subtipos histológicos de la IWF (International Working Formulation) B, C y D si se compara con el subtipo IWF A (58 % vs 12 %), mayor en pacientes en los que el diámetro mayor de la lesión más grande era < 5 cm vs > 7 cm (53 % vs 38 %) y mayor, también, en pacientes con recidiva quimiosensible si se compara con las quimioresistentes (definidas como duración de la respuesta < 3 meses) (50 % vs 22 %). La TRG en pacientes sometidos previamente a un trasplante autólogo de médula ósea (TAMO) fue del 78 % vs. 43 % en pacientes sin TAMO. Ni factores como la edad, sexo, grado del linfoma, diagnóstico inicial, presencia o ausencia de enfermedad bulky, LDH normal o elevada ni la presencia de enfermedad extranodal tuvieron un efecto estadísticamente significativo (test de exactitud de Fisher) sobre la respuesta de rituximab. Se ha determinado una correlación estadísticamente significativa entre la tasa de respuesta y la afectación de la médula ósea. El 40 % de los pacientes con afectación de la médula ósea respondieron al tratamiento frente al 59 % de los pacientes sin afectación de la médula ósea (p=0,0186). Este hallazgo no fue apoyado por un análisis de regresión logística por etapas en el cual se identificaron los siguientes factores pronósticos: tipo histológico, positividad basal para bcl-2, resistencia a la última quimioterapia administrada y enfermedad bulky.
Tratamiento inicial, semanal, 8 dosis
En un ensayo multicéntrico, de un único brazo, 37 pacientes con LNH recidivante o quimioresistente, de bajo grado o folicular de células B recibieron 375 mg/m2 de rituximab como perfusión intravenosa semanal durante 8 dosis. La TRG fue del 57 % (Intervalo de confianza (IC) del 95 %; 41 % - 73 %; RC 14 %, RP 43 %) con una mediana proyectada del TTP de 19,4 meses (rango 5,3 a 38,9 meses) para los pacientes que respondieron a la terapia.
Tratamiento inicial, enfermedad bulky, semanal, 4 dosis
Con datos agrupados de tres ensayos clínicos, 39 pacientes con LNH de tipo bulky recidivante o quimioresistente, de bajo grado o folicular de células B(lesión única ≥ 10 cm de diámetro) recibieron 375 mg/m2 de rituximab como perfusión intravenosa semanal durante 4 dosis. La TRG fue del 36 % (IC95 % 21 % - 51 %; RC 3 %, RP 33 %) con una mediana del TTP de 9,6 meses (rango 4,5 a 26,8 meses) para los pacientes que respondieron a la terapia.
Retratamiento, semanal, 4 semanas
En un ensayo multicéntrico, de un único brazo, 58 pacientes con LNH en recidiva o quimioresistente, de bajo grado o folicular de células B, que habían alcanzado una respuesta clínica objetiva en un tratamiento previo con rituximab, fueron re-tratados con 375 mg/m2 de rituximab en perfusión intravenosa semanal durante cuatro semanas. Tres de estos pacientes habían recibido dos ciclos de tratamiento antes de ser incluidos en el ensayo, lo que significa que recibieron su tercer ciclo de tratamiento en el ensayo. Dos pacientes fueron re-tratados dos veces en el ensayo. En los 60 retratamientos del ensayo, la TRG fue del 38 % (IC95 % 26 % - 51 %; RC 10 %, RP 28 %) con una mediana proyectada de TTP de 17,8 meses (rango 5,4 a 26,6 meses) para los pacientes que respondedores. Estos datos son más favorables que el TTP alcanzado tras el tratamiento previo con Rituximab (12,4 meses).
Tratamiento inicial, en combinación con quimioterapia
En un ensayo abierto aleatorizado, un total de 322 pacientes con linfoma folicular no tratados previamente recibieron quimioterapia CVP (ciclofosfamida 750 mg/m2, vincristina 1,4 mg/m2 hasta un máximo de 2 mg el día 1, y prednisolona 40 mg/m2/día los días 1-5) cada 3 semanas durante 8 ciclos o bien Rituximab 375 mg/m2 en combinación con CVP (R-CVP). Rituximab se administró el primer día de cada ciclo de tratamiento. La eficacia se evaluó en un total de 321 pacientes tratados (162 R-CVP, 159 CVP). La mediana de la duración del seguimiento de los pacientes fue de 53 meses. El grupo tratado con R-CVP obtuvo un beneficio significativo sobre el tratado con CVP en lo que respecta a la variable principal de eficacia, el tiempo hasta el fracaso del tratamiento (TFT) (27 meses vs. 6,6 meses; p < 0,0001. log-rank test). La proporción de pacientes con respuesta antitumoral (RC, respuesta completa no confirmada RCnc, RP) fue significativamente superior (p < 0,0001 test Chi-cuadrado) en el grupo R-CVP (80,9 %) que en el grupo CVP (57,2 %). El tratamiento R-CVP prolongó significativamente el tiempo hasta progresión de la enfermedad o fallecimiento en comparación con CVP; 33,6 meses y 14,7 meses, respectivamente (p < 0,0001, log-rank test). La mediana de la duración de la respuesta fue de 37,7 meses en el grupo R-CVP y de 13,5 meses en el grupo CVP (p < 0,0001, log-rank test).
La diferencia entre los grupos de tratamiento con respecto a la supervivencia global fue clínicamente significativa (p = 0,029; log-rank test estratificado por centros): la tasa de supervivencia a los 53 meses fue del 80,9 % para pacientes del grupo R-CVP comparada con el 71,1 % para pacientes del grupo CVP.
Los resultados de otros tres ensayos aleatorizados en los que se utilizó rituximab en combinación con un régimen de quimioterapia distinto de CVP (CHOP, MCP, CHVP/Interferón-α) han demostrado también mejorías significativas en las tasas de respuesta, en parámetros dependientes del tiempo, así como en supervivencia global. La Tabla 8 resume los resultados clave de los cuatro ensayos.
Tabla 8 Resumen de los resultados clave de cuatro ensayos aleatorizados en fase III que evaluaron el beneficio de rituximab con diferentes regímenes de quimioterapia en linfoma folicular
| Estudio | Tratamiento, N | Mediana de la duración del tratamiento, meses | Tasa de respuesta global (TRG), % | Respuesta completa (RC), % | Mediana del TTP/SLP/SLE, meses | Tasas SG, % |
| M39021 | CVP, 159 R-CVP, 162 | 53 | 57 81 | 10 41 | Mediana del TTP: 14,7 33,6 p < 0,0001 | 53 meses 71,1 80,9 p = 0,029 |
| GLSG’00 | CHOP, 205 R-CHOP, 223 | 18 | 90 96 | 17 20 | Mediana del TFT: 2,6 años No alcanzado p < 0,001 | 18 meses 90 95 p = 0,016 |
| OSHO-39 | MCP, 96 R-MCP, 105 | 47 | 75 92 | 25 50 | Mediana de la SLP 28,8 No alcanzado p < 0,0001 | 48 meses 74 87 p = 0,0096 |
| FL2000 | CHVP-IFN, 183 R-CHVP-IF N, 175 | 42 | 85 94 | 49 76 | Mediana de la SLE: 36 No alcanzado p < 0,0001 | 42 meses 84 91 p = 0,029 |
SLE – Supervivencia libre de evento
TTP – Tiempo hasta la progresión o la muerte SLP – Supervivencia libre de progresión
TFT – Tiempo hasta el fallo del tratamiento
Tasas de SG – tasas de supervivencia en el momento de los análisis
SLE – Supervivencia libre de evento
TTP – Tiempo hasta la progresión o la muerte SLP – Supervivencia libre de progresión
TFT – Tiempo hasta el fallo del tratamiento
Tasas de SG – tasas de supervivencia en el momento de los análisis
Terapia de mantenimiento
Linfoma folicular previamente no tratado
En un ensayo de fase III, prospectivo, abierto, internacional y multicéntrico, a 1193 pacientes con linfoma folicular avanzado no tratados previamente se les administró terapia de inducción con RCHOP (n= 881); R-CVP (n= 268) o R-FCM (n=44) a criterio del investigador. Un total de 1078 respondieron a la terapia de inducción, de los cuales 1018 fueron aleatorizados a terapia de mantenimiento con rituximab (n=505) u observación (n=513). Los dos grupos de tratamiento estuvieron bien equilibrados en cuanto a las características basales y el estado de la enfermedad. El tratamiento de mantenimiento consistió en una perfusión de rituximab 375mg/m2 de superficie corporal administrada cada 2 meses hasta progresión de la enfermedad o durante un periodo máximo de 2 años.
El análisis primario predeterminado se llevó a cabo en una mediana de tiempo de observación de 25 meses desde la aleatorización, la terapia de mantenimiento con rituximab arrojó resultados clínicamente relevantes y estadísticamente significativos en la variable principal Supervivencia libre de progresión evaluada por el investigador (SLP), comparado con los pacientes en observación con linfoma folicular previamente no tratado (Tabla 9).
También se observó beneficio significativo del mantenimiento con rituximab en las variables secundarias del análisis primario: supervivencia libre de evento (SLE), tiempo hasta nuevo tratamiento del linfoma (TNLT), tiempo hasta nuevo tratamiento de quimioterapia (TNCT) y tasa de respuesta global (TRG/ORR) (Tabla 9).
Los datos del seguimiento de los pacientes en el estudio (mediana de seguimiento de 9 años) confirmaron el beneficio a largo plazo de la terapia de mantenimiento con rituximab en términos de SLP, SLE, TNLT y TNCT (Tabla 9).
Tabla 9 Resumen de los resultados de eficacia para el tratamiento de mantenimiento con rituximab frente a observación en el análisis principal definido por el protocolo y después de una mediana de seguimiento de 9 años (análisis final)
| Análisis principal (mediana del S: 25 meses) | Análisis final (mediana del S: 9,0 años) | |||
| Rituximab N = 505 | Rituximab N = 505 | |||
| Supervivencia libre de progresión (mediana) valor p-logarítmico ordinal cociente de riesgo (IC del 95 %) reducción del riesgo | ||||
| < 0,0001 0,50 (0,39; 0,64) 50 % | < 0,0001 0,61 (0,52; 0,73) 39 % | |||
| Supervivencia global (mediana) valor p-logarítmico ordinal cociente de riesgo (IC del 95 %) reducción del riesgo | ||||
| 0,7246 0,89 (0,45; 1,74) 11 % | 0,7948 1,04 (0,77; 1,40) -6 % | |||
| valor p-logarítmico ordinal cociente de riesgo (IC del 95 %) reducción del riesgo | < 0,0001 0,54 (0,43; 0,69) 46 % | < 0,0001 0,64 (0,54; 0,76) 36 % | ||
| valor p-logarítmico ordinal cociente de riesgo (IC del 95 %) reducción del riesgo | 0,0003 0,61 (0,46; 0,80) 39 % | < 0,0001 0,66 (0,55; 0,78) 34 % | ||
| valor p-logarítmico ordinal cociente de riesgo (IC del 95 %) reducción del riesgo | 0,0011 0,60 (0,44; 0,82) 40 % | 0,0004 0,71 (0,59; 0,86) 39 % | ||
| valor p de la prueba de la χ2 razón de posibilidades (IC del 95 %) | < 0,0001 2,33 (1,73; 3,15) | < 0,0001 2,43 (1,84; 3,22) | ||
| valor p de la prueba de la χ2 razón de posibilidades (IC del 95 %) | < 0,0001 2,21 (1,65; 2,94) | < 0,0001 2,34 (1,80; 3,03) | ||
* al final del mantenimiento/observación; resultados del análisis final según la mediana de seguimiento de 73 meses. S: seguimiento; NA: no alcanzado en la fecha de corte de datos clínicos; TNLT: tiempo hasta el próximo tratamiento antilinfoma; TNCT: tiempo hasta la próxima quimioterapia.
El tratamiento de mantenimiento con rituximab ha supuesto un beneficio en todos los subgrupos predefinidos evaluados: sexo (hombre, mujer), edad (< 60 años, ≥60 años), escala FLIPI (≤1,2 o ≥3), terapia de inducción (R-CHOP, R-CVP, R-FCM), y sin tener en cuenta la calidad de la respuesta al tratamiento de inducción (CRu, CR o RP). Análisis exploratorios sobre el beneficio del tratamiento de mantenimiento, mostraron un efecto menos pronunciado en pacientes de edad avanzada (> 70 años), sin embargo, los tamaños de muestra fueron pequeños.
Linfoma folicular recidivante/refractario
En un ensayo de fase III, prospectivo, abierto, internacional y multicéntrico, 465 pacientes con linfoma folicular en recidiva o refractario se randomizaron aleatoriamente en la primera fase para recibir una terapia de inducción con CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona; n=231) o con rituximab más CHOP (R-CHOP, n=234). Los dos grupos de tratamiento estuvieron bien equilibrados en cuanto a las características basales y el estado de la enfermedad. En la segunda fase los 334 pacientes que consiguieron una remisión completa o parcial tras la terapia de inducción fueron distribuidos aleatoriamente, para recibir terapia de mantenimiento con rituximab (n=167) u observación (n=167). El tratamiento de mantenimiento con rituximab consistió en una perfusión única de rituximab a una dosis de 375 mg/m2 de superficie corporal, administrado cada 3 meses hasta progresión de la enfermedad o durante un periodo máximo de 2 años.
El análisis de eficacia final incluyó a todos los pacientes aleatorizados en ambas fases del ensayo. Tras una mediana de tiempo de observación de 31 meses, el resultado de los pacientes con linfoma folicular en recidiva o refractario incluidos en la fase de inducción, con R-CHOP mejoró significativamente comparado con CHOP (ver Tabla 10).
Tabla 10 Fase de inducción: resumen de los resultados de eficacia para CHOP frente a R-CHOP (mediana del tiempo de observación de 31 meses)
| CHOP | R-CHOP | Valor p |
| 58 % | 58 % | 0,9449 |
1) Las estimaciones se calcularon por cocientes de riesgo.
2) Última respuesta tumoral según la evaluación del investigador. La prueba estadística “principal” para “respuesta” fue un test de tendencia de RC frente a RP frente a no respuesta (p < 0,0001)
Abreviaturas: ND, no disponible; TRG: tasa de respuesta global; RC: respuesta completa; RP: respuesta parcial
Para los pacientes aleatorizados en la fase de mantenimiento del estudio, la mediana del tiempo de observación fue 28 meses desde la aleatorización. El tratamiento de mantenimiento con rituximab condujo a una mejoría significativa y clínicamente relevante de la variable principal de eficacia, la supervivencia libre de progresión o SLP (tiempo desde la aleatorización al tratamiento de mantenimiento hasta la recidiva, progresión de la enfermedad o fallecimiento) en comparación con los pacientes en observación (p< 0,0001 log-rank test). La mediana de la SLP fue 42,2 meses en el brazo de mantenimiento con rituximab en comparación con los 14,3 meses del brazo de observación. Utilizando el análisis de regresión de Cox, el riesgo de experimentar una progresión de la enfermedad o fallecimiento se redujo un 61 % con el tratamiento de mantenimiento con rituximab en comparación con el brazo de observación (95 % IC; 45 %-72 %). Según una estimación de las curvas de Kaplan–Meier, la supervivencia libre de eventos a los 12 meses fue de 78 % en el grupo de mantenimiento con rituximab vs 57 % en el grupo en observación. Un análisis de la supervivencia global confirmó el beneficio significativo del tratamiento de mantenimiento con rituximab versus observación (p=0,0039 log-rank test). El tratamiento de mantenimiento con rituximab redujo el riesgo de fallecimiento en un 56 % (95 % IC: 22 %-75 %).
Tabla 11 Fase de mantenimiento: resumen de los resultados de eficacia de rituximab frente a observación (mediana del tiempo de observación 28 meses)
| Variable de eficacia | Estimación de Kaplan-Meier de la mediana del tiempo hasta evento (meses) | Reducción del riesgo | ||
| Observación (N = 167) | Rituximab (N = 167) | Valor p del orden logarítmico | ||
| Supervivencia libre de progresión (SLP) | 14,3 | 42,2 | < 0,0001 | 61 % |
| Supervivencia global | NA | NA | 0,0039 | 56 % |
| Tiempo para el nuevo tratamiento de linfoma | 20,1 | 38,8 | < 0,0001 | 50 % |
| Supervivencia libre de enfermedada | 16,5 | 53,7 | 0,0003 | 67 % |
| R-CHOP | NA | NA | 0,0482 | 56 % |
NA: no alcanzado; a: únicamente aplicable a los pacientes que alcanzaron una RC
El beneficio del tratamiento de mantenimiento con rituximab se confirmó en todos los subgrupos analizados, independientemente del régimen de inducción (CHOP o R-CHOP) o la calidad de la respuesta al tratamiento de inducción (RC o RP) (Tabla 11). El tratamiento de mantenimiento con rituximab prolongó significativamente la mediana de la SLP en los pacientes respondedores a la terapia de inducción con CHOP (mediana de SLP 37,5 meses vs 11,6 meses, p< 0,0001) y también en los pacientes respondedores a la terapia de inducción con R-CHOP (mediana de SLP 51,9 meses vs 22,1 meses, p=0,0071). Aunque los subgrupos fueron pequeños, el tratamiento de mantenimiento con rituximab proporcionó un beneficio significativo en términos de supervivencia global tanto para los pacientes respondedores a CHOP como para los pacientes respondedores a R-CHOP, aunque se necesita un seguimiento más largo para confirmar esta observación.
Linfoma no Hodgkin difuso de células B grandes en adultos
En un ensayo abierto, aleatorizado, un total de 399 pacientes de edad avanzada previamente no tratados (edad entre 60 y 80 años) que padecían linfoma difuso de células B grandes recibieron quimioterapia CHOP estándar (ciclofosfamida 750 mg/m2, doxorrubicina 50 mg/m2, vincristina 1,4 mg/m2 hasta un máximo de 2 mg el día 1 y prednisolona 40 mg/m2/día los días 1-5) cada 3 semanas durante 8 ciclos o bien recibieron rituximab 375 mg/m2 más CHOP (R-CHOP).
Rituximab se administró el primer día de cada ciclo de tratamiento.
El análisis final de eficacia incluyó todos los pacientes aleatorizados (197 CHOP, 202 R-CHOP), y obtuvo una mediana de tiempo de seguimiento de aproximadamente 31 meses. Los dos grupos de tratamiento estuvieron bien equilibrados en cuanto a las características basales y el estatus de la enfermedad. El análisis final confirmó que el tratamiento con R-CHOP estaba asociado con una mejoría clínicamente relevante y estadísticamente significativa en la duración de la supervivencia libre de evento (variable principal de eficacia, donde los eventos fueron fallecimiento, recidiva o progresión del linfoma o instauración de un nuevo tratamiento anti-linfoma) (p=0,0001). La estimación de la duración media de la supervivencia libre de evento fue de 35 meses según las curvas de Kaplan Meier en el brazo tratado con R-CHOP frente a 13 meses en el brazo con CHOP, lo cual representa una reducción del riesgo del 41 %. A 24 meses, la estimación de la supervivencia global fue del 68,2 % en el brazo con R-CHOP frente al 57,4 % en el brazo con CHOP. Un análisis posterior de la duración de la supervivencia global, durante un periodo de seguimiento de 60 meses de mediana, confirmó el beneficio del tratamiento de R-CHOP sobre el CHOP (p=0,0071), lo cual representa una reducción del riesgo del 32 %.
El análisis de todas las variables secundarias (índices de respuesta, supervivencia libre de progresión, supervivencia libre de enfermedad, duración de la respuesta) confirma el efecto del tratamiento con R - CHOP cuando se compara con CHOP. El índice de respuestas completas tras el ciclo 8 fue de 76,2 % en el grupo con R-CHOP y de 62,4 % en el grupo con CHOP (p=0,0028). El riesgo de progresión de la enfermedad se redujo en un 46 % y el riesgo de recidiva en un 51 %. En todos los subgrupos de pacientes (sexo, edad, IPI ajustado por edad, estadio Ann Arbor, ECOG, β2 microglobulina, LDH, albúmina, síntomas- B, enfermedad bulky, localizaciones extranodulares, afectación de médula ósea), las relaciones de riesgo para la supervivencia libre de evento y la supervivencia global (R-CHOP comparado con CHOP) fueron menos de 0,83 y 0,95, respectivamente. El R-CHOP se asoció con mejoría en el resultado del tratamiento tanto de pacientes de alto como de bajo riesgo, de acuerdo al IPI ajustado por edad.
Hallazgos clínicos de laboratorio
No se apreciaron respuestas en ninguno de los 67 pacientes en los que se evaluó la presencia de anticuerpos anti-murino humano (HAMA). De los 356 pacientes en los que se evaluó la presencia de anticuerpos antifármaco (ADA), el 1,1 % fueron positivos (4 pacientes).
Leucemia linfocítica crónica
En dos ensayos randomizados y abiertos, un total de 817 pacientes con leucemia linfática crónica no tratados previamente y 552 pacientes con LLC en recidiva o refractaria se aleatorizaron para recibir cada uno quimioterapia FC (fludarabina 25 mg/m2, ciclofosfamida 250 mg/m2, los días 1-3) cada 4 semanas durante 6 ciclos o rituximab en combinación con FC (R-FC). Rituximab se administró a una dosis de 375 mg/m2 un día antes de la quimioterapia durante el primer ciclo y a una dosis de 500 mg/m2 en el día 1 de cada ciclo de tratamiento posterior. Los pacientes fueron excluidos del estudio de LLC en recidiva o refractaria si previamente habían sido tratados con anticuerpos monoclonales o si eran refractarios a fludarabina o a algún otro análogo de nucleósido (definido como fracaso para alcanzar una remisión parcial durante al menos 6 meses). Se analizaron para la eficacia un total de 810 pacientes (403 R-FC, 407 FC) para estudios de primera línea (Tabla 12a y Tabla 12b) y 552 pacientes (276 R-FC, 276 FC) para el estudio de recidiva o refractaria (Tabla 13).
En los estudios en primera línea, después de una mediana del tiempo de observación de 48,1 meses, la mediana de SLP fue de 55 meses en el grupo R-FC y 33 meses en el grupo FC (p <0,0001, log-rank test). El análisis de supervivencia global mostró un beneficio significativo del tratamiento con R-FC frente a FC, quimioterapia sola, (p = 0.0319, log-rank test) (Tabla 12a). El beneficio en términos de SLP se observó consistentemente en muchos de los subgrupos de pacientes analizados de acuerdo al riesgo de enfermedad de base (es decir, clasificación de Binet A-C) (Tabla 12b).
Tabla 12a Tratamiento de primera línea de la leucemia linfocítica crónica
Resumen de los resultados de eficacia para rituximab con FC frente a FC sola - mediana de tiempo de observación de 48,1 meses
| Parámetro de eficacia | Estimación de Kaplan-Meier de la mediana del tiempo hasta evento (meses) | Reducción del riesgo | ||
| FC (N = 409) | R-FC (N = 408) | Valor p-logarítmico ordinal | ||
| Supervivencia libre de progresión (SLP) | 32,8 | 55,3 | < 0,0001 | 45 % |
| Supervivencia global | NA | NA | 0,0319 | 27 % |
| Supervivencia libre de evento | 31,3 | 51,8 | < 0,0001 | 44 % |
| Tasa de respuesta (RC, RPn o RP) Tasas de RC | 72,6 % 16,9 % | 85,8 % 36,0 % | < 0,0001 < 0,0001 | NP NP |
| Duración de la respuesta* | 36,2 | 57,3 | < 0,0001 | 44 % |
| Supervivencia libre de enfermedad (SLE)** | 48,9 | 60,3 | 0,0520 | 31 % |
| Tiempo hasta el nuevo tratamiento | 47,2 | 69,7 | < 0,0001 | 42 % |
Tasa de respuesta y tasas de RC analizadas mediante la prueba de la χ2. NA: no alcanzado; NP: no procede
*: únicamente aplicable a pacientes que alcanzaron una RC, RPn, RP
**: únicamente aplicable a pacientes que alcanzaron una RC
Tabla 12b Tratamiento de primera línea de la leucemia linfocítica crónica
Cocientes de riesgo de supervivencia libre de progresión según el estadio de Binet (ITT)–mediana de tiempo de observación de 48,1 meses
| Supervivencia libre de progresión (SLP) | Número de pacientes | Cociente de riesgo (IC del 95 %) | Valor p (Prueba de Wald, no ajustada) | |
| FC | R-FC | |||
| Estadio A de Binet | 22 | 18 | 0,39 (0,15; 0,98) | 0,0442 |
| Estadio B de Binet | 259 | 263 | 0,52 (0,41; 0,66) | < 0,0001 |
| Estadio C de Binet | 126 | 126 | 0,68 (0,49; 0,95) | 0,0224 |
IC: Intervalo de confianza
En el estudio de LLC recidivante/refractaria, la mediana de supervivencia libre de progresión (variable principal) fue de 30,6 meses en el grupo R-FC y de 20,6 meses en el grupo FC (p = 0,0002, log-rank test). El beneficio en términos de SLP se observó en casi todos los subgrupos de pacientes analizados según el riesgo de enfermedad de base. Un ligero, pero no significativo aumento en la supervivencia global fue notificado en la comparación del R-FC con el brazo FC.
Tabla 13 Tratamiento de la leucemia linfocítica crónica
recidivante/refractaria - resumen de los resultados de eficacia para rituximab con FC frente a FC sola (mediana del tiempo de observación de 25,3 meses)
| Parámetro de eficacia | Estimación de Kaplan-Meier de la mediana del tiempo hasta evento(meses) | Reducción del riesgo | ||
| FC (N = 276) | R-FC (N = 276) | Valor p-logarítmico ordinal | ||
| Supervivencia libre de progresión (SLP) | 20,6 | 30,6 | 0,0002 | 35 % |
| Supervivencia global | 51,9 | NA | 0,2874 | 17 % |
| Supervivencia libre de evento | 19,3 | 28,7 | 0,0002 | 36 % |
| Supervivencia libre de enfermedad (SLE)** Tiempo para un nuevo tratamiento de LLC | 34,2 | NA | 0,0024 | 35 % |
Tasa de respuesta y tasas de RC analizadas mediante la prueba de la χ2.
*: únicamente aplicable a pacientes que alcanzaron una RC, RPn, RP; NA: no alcanzado NP: no procede
**: únicamente aplicable a pacientes que alcanzaron una RC.
Los resultados de otros estudios de soporte que utilizaron rituximab en combinación con otros regímenes de quimioterapia (incluido CHOP, FCM, PC, PCM, bendamustina y cladribina) para el tratamiento de pacientes con leucemia linfática crónica (LLC) en recidiva o refractaria o no tratados previamente demostraron también una alta tasa de respuesta global con beneficios en términos de SLP, aunque con una toxicidad ligeramente más alta (especialmente mielotoxicidad). Estos estudios apoyan el uso de rituximab con otra quimioterapia.
Los datos en aproximadamente 180 pacientes tratados previamente con rituximab han demostrado un beneficio clínico (incluyendo respuesta completa RC) y avalan el retratamiento con rituximab.
Población pediátrica
Se realizó un estudio abierto, aleatorizado y multicéntrico de quimioterapia LMB (corticosteroides, vincristina, ciclofosfamida, dosis altas de metotrexato, citarabina, doxorrubicina, etopósido y terapia intratecal triple fármaco [metotrexato/citarabina/corticosteroide]) solo o en combinación con rituximab en pacientes pediátricos con estadio avanzado de LBDCG CD20 positivo/LB/LLA-B maduro/LBL previamente no tratados. El estadio avanzado se define como el Estadio III con un nivel elevado de LDH (“B-alto”), [LDH> dos veces el límite superior de los valores normales para adultos (> Nx2)] o cualquier estadio IV o LLA-B. Los pacientes fueron aleatorizados para recibir quimioterapia LMB o seis perfusiones IV de rituximab a una dosis de 375 mg/m2 de superficie corporal en combinación con quimioterapia LMB (dos durante cada uno de los dos ciclos de inducción y uno durante cada uno de los dos ciclos de consolidación) según el esquema Lymphome Malin B. Se incluyeron un total de 328 pacientes aleatorizados en los análisis de eficacia, de los cuales un paciente menor de 3 años recibió rituximab en combinación con quimioterapia LMB.
Los dos brazos de tratamiento, LMB (quimioterapia LMB) y R-LMB (quimioterapia LMB con rituximab), estaban bien equilibrados con respecto a las características basales. Los pacientes tenían una edad media de 7 y 8 años en el brazo LMB y el brazo R-LMB, respectivamente.
Aproximadamente la mitad de los pacientes estaban en el Grupo B (50,6 % en el brazo LMB y 49,4 % en el brazo R-LMB), 39,6 % en el Grupo C1 en ambos brazos, y 9,8 % y 11,0 % estaban en el Grupo C3 en los brazos LMB y R-LMB, respectivamente. Según la estadificación de Murphy, la mayoría de los pacientes eran LB estadio III (45,7 % en el brazo LMB y 43,3 % en el brazo R-LMB) o LLA-B madura negativo en SNC (21,3 % en el brazo LMB y 24,4 % en el brazo R-LMB). Menos de la mitad de los pacientes (45,1 % en ambos brazos) tenían afectación de la médula ósea, y la mayoría de los pacientes (72,6 % en el brazo LMB y 73,2 % en el brazo R-LMB) no tenían afectación del SNC. El criterio de valoración principal de eficacia fue supervivencia libre de evento (SLE), donde un evento se definió como la aparición de enfermedad progresiva, recaída, segunda neoplasia maligna, muerte por cualquier causa o falta de respuesta demostrada mediante la detección de células viables en masas residuales después del segundo ciclo CYVE, lo que ocurra primero. Los criterios de valoración secundarios de eficacia fueron supervivencia global (SG) y remisión completa (RC).
En el análisis intermedio preespecificado con aproximadamente 1 año de mediana de seguimiento, se observó una mejoría clínicamente relevante en el criterio de valoración principal de SLE, con estimaciones de tasas a 1 año del 94,2 % (IC 95 %, 88,5 % – 97,2 %) en el brazo R-LMB vs. 81,5 % (IC 95 %, 73,0 % - 87,8 %) en el brazo LMB, y un Cox HR ajustado de 0,33 (IC 95 %, 0,14 – 0,79). Por recomendación del IDMC (comité independiente de monitorización de datos) basada en este resultado, la aleatorización se detuvo y los pacientes en el brazo LMB pudieron cambiarse para recibir rituximab.
Los análisis de eficacia finales se realizaron en 328 pacientes aleatorizados con una mediana de seguimiento de 3,1 años. Los resultados se describen en la Tabla 14.
Tabla 14 Resumen de los resultados de eficacia finales (población ITT)
| Análisis | LMB (N = 164) | R-LMB (N = 164) |
| SLE | 28 eventos | 10 eventos |
| Long-rank test unilateral con un p-valor de 0,0006 | ||
| Cox HR ajustado 0,32 (IC 90 %: 0,17; 0,58) | ||
| SLE en 3 años | 82,3 % (IC 95 %: 75,7 %, 87,5 %) | 93,9 % (IC 95 %: 89,1%, 96,7%) |
| SG | 20 muertes | 8 muertes |
| Long-rank test unilateral con un p-valor de 0,0061 | ||
| Cox HR ajustado 0,36 (IC 95 %: 0,16; 0,81) | ||
| SG en 3 años | 87,3 % (IC 95 %: 81,2 %; 91,6 %) | 95,1 % (IC 95 %: 90,5 %; 97,5 %) |
| Tasa RC | 93,6 % (95 %: 88,2 %; 97,0 %) | 94,0 % (IC 95 %: 88,8 %; 97,2 %) |
Abreviaturas: SLE: supervivencia libre de eventos; SG: supervivencia global; CR: remisión completa
El análisis de eficacia primario mostró un beneficio en SLE de la adición de rituximab a la quimioterapia LMB frente a la quimioterapia LMB sola, con un HR de SLE de 0,32 (IC 90 % 0,17 – 0,58) según un análisis de regresión de Cox que se ajusta a la nacionalidad, a la histología y al grupo terapéutico. Mientras que no se observaron mayores diferencias en el número de pacientes que alcanzaron remisión completa entre los dos grupos de tratamiento, el beneficio de la adición de rituximab a la quimioterapia LMB también se mostró en el criterio de valoración secundario de SG con un HR de 0,36 (IC 95 %, 0,16 – 0,81).
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con rituximab en todos los grupos de la población pediátrica con linfoma folicular y LLC, y en la población pediátrica desde el nacimiento hasta < 6 meses de edad en el linfoma difuso de células B grandes CD20 positivo. Ver sección 4.2 para consultar la información sobre el uso en la población pediátrica.
Experiencia clínica en artritis reumatoide
La eficacia y seguridad de rituximab para el alivio de los síntomas y signos de la artritis reumatoide en pacientes con una respuesta inadecuada a los inhibidores del TNF se demostró en un ensayo pivotal, aleatorizado, controlado, doble ciego y multicéntrico (ensayo 1).
El ensayo 1 evaluó a 517 pacientes que habían experimentado una respuesta inadecuada o intolerancia a uno o más tratamientos inhibidores de TNF. Los pacientes aptos sufrían artritis reumatoide activa, diagnosticada según los criterios del Colegio Estadounidense de Reumatología (ACR, por sus siglas en inglés). Rituximab se administró como dos perfusiones IV separadas por un intervalo de 15 días. Los pacientes recibieron 2 perfusiones intravenosas de 1.000 mg de rituximab o placebo en combinación con MTX. Todos los pacientes recibieron 60 mg de prednisona oral concomitante los días 2-7 y 30 mg los días 8-14 después de la primera perfusión. La variable principal de eficacia fue la proporción de pacientes que lograron una respuesta ACR20 a las 24 semanas. Se realizó un seguimiento de los pacientes durante más de 24 semanas para los objetivos a largo plazo, incluida la valoración radiográfica a las 56 semanas y a las 104 semanas. Durante este tiempo, el 81 % de los pacientes, del grupo placebo original, recibieron rituximab entre las semanas 24 y 56, conforme a un protocolo abierto de extensión del ensayo.
Ensayos de rituximab en pacientes con artritis temprana (pacientes sin tratamiento previo con metotrexato y pacientes con una respuesta inadecuada a metotrexato, pero aún no tratados con inhibidores de TNF-alfa) han alcanzado sus objetivos primarios. Rituximab no está indicado en estos pacientes, ya que los datos de seguridad sobre el tratamiento de rituximab a largo plazo son insuficientes, en particular, sobre el riesgo de desarrollo de tumores malignos y LMP.
Resultados de la actividad de la enfermedad
Rituximab en combinación con metotrexato aumentó significativamente el porcentaje de pacientes con una mejora mínima del 20 % en la puntuación ACR, en comparación con los tratados únicamente con metotrexato (Tabla 15). En todos los ensayos de desarrollo el beneficio terapéutico resultó similar para los pacientes, al margen de edad, género, superficie corporal, raza, número de tratamientos anteriores o estado de la enfermedad.
Asimismo, se advirtió una mejoría clínica y estadísticamente significativa de cada uno de los componentes de la respuesta ACR (número de articulaciones dolorosas y tumefactas), evaluación general del paciente y del médico, puntuaciones de los índices de discapacidad (HAQ), evaluación del dolor y Proteína C Reactiva (mg/dL).
Tabla 15 Resultados de la respuesta clínica de la variable principal en el ensayo 1 (población por ITT)
| Resultado† | Placebo + MTX | Rituximab + MTX (2 x 1.000 mg) | |
| Ensayo 1 | N = 201 | N = 298 | |
| ACR20 ACR50 ACR70 | 36 (18 %) 11 (5 %) 3 (1 %) | 153 (51 %)*** 80 (27 %)*** 37 (12 %)*** | |
| Respuesta EULAR (Buena/Moderada) | 44 (22 %) | 193 (65 %)*** | |
| Diferencia media en DAS | –0,34 | –1,83*** |
Resultado † a las 24 semanas
Diferencia significativa de placebo + MTX en el punto temporal principal: ***p ≤ 0,0001
Los pacientes tratados con rituximab en combinación con metotrexato experimentaron un descenso significativamente mayor de la actividad de la enfermedad según la escala (DAS28) que los tratados únicamente con metotrexato (Tabla 15). De igual forma, se obtuvo una respuesta EULAR (European League Against Rheumatism) de buena a moderada significativamente mejor en los pacientes tratados con rituximab más metotrexato que en los pacientes tratados con metotrexato sólo. (Tabla 15).
Respuesta radiográfica
El daño estructural articular fue valorado radiográficamente y expresado como el cambio en el Índice Total de Sharp modificado (ITSm) y sus componentes, el índice de erosión y el índice de estrechamiento del espacio articular.
En el ensayo 1 en pacientes con respuesta inadecuada o intolerancia a uno o más tratamientos inhibidores de TNF, que recibieron rituximab en combinación de metotrexato demostraron una progresión radiográfica significativamente menor que los pacientes que inicialmente recibieron metotrexato solo durante 56 semanas. De los pacientes que inicialmente recibieron sólo metotrexato, el 81 % recibieron rituximab como rescate entre las semanas 16-24, o en el estudio de extensión antes de la semana 56. Una alta proporción de los pacientes que recibieron inicialmente el tratamiento de rituximab /MTX no tuvieron progresión en el índice de erosión en la semana 56 (Tabla 16).
Tabla 16 Resultados radiográficos a 1 año (población por ITSm)
| Placebo + MTX | Rituximab + MTX 2 x 1.000 mg | |
| Ensayo 1 | (N = 184) | (N = 273) |
| Proporción de pacientes sin cambio erosivo | 52 % | 60 %, NS |
150 pacientes aleatorizados originalmente a placebo + MTX en el ensayo 1 recibieron al menos un ciclo de RTX + MTX al año.
* p < 0,05, ** p < 0,001. Abreviatura: NS, no significativo
La inhibición de la progresión del daño articular se observó también a largo plazo. En el ensayo 1, el análisis radiográfico a 2 años demostró una disminución significativa de la progresión del daño estructural articular en los pacientes que recibieron rituximab en combinación con metotrexato comparado con metotrexato sólo, así como un aumento significativo de la proporción de pacientes sin progresión del daño articular durante un periodo de dos años.
Función física y resultados de calidad de vida
En los pacientes tratados con rituximab se observaron descensos significativos del índice de discapacidad (HAQ-DI) y del índice de fatiga (FACIT-Fatiga) en comparación con los que sólo habían recibido metotrexato La proporción de pacientes tratados con rituximab que mostraban una mínima progresión clínicamente importante (MPCI) en HAQ-DI (definida por un descenso de la puntuación total individual de > 0,22) fue mayor que en los pacientes que recibieron sólo metotrexato (Tabla 17).
Se demostró también una mejora significativa de la calidad de vida relativa a la salud con la mejora significativa del índice de salud física (ISF) y del índice de salud mental (ISM) de SF-36. Además, un porcentaje significativamente mayor logró MPCI en estos índices (Tabla 17)
Tabla 17 Función física y resultados de la calidad de vida en el Ensayo 1, en la semana 24
| Resultado† | Placebo + MTX | Rituximab + MTX (2 x 1.000 mg) |
| % SF-36 MHS MPCI | 20 % | 38 %* |
Resultado † a las 24 semanas
Diferencia significativa con respecto al placebo en el punto temporal principal: * p < 0,05, ** p < 0,001 *** p ≤ 0,0001
MPCI HAQ- DI ≥ 0.22, MPCI SF-36 PSH > 5.42, MPCI SF-36 PSM > 6.33
Eficacia en pacientes seropositivos a autoanticuerpos (FR y/o anti-CCP)
Los pacientes seropositivos al Factor Reumatoide (FR) y/o anti-Péptido-Cíclico Citrulinado (anti-CCP) que fueron tratados con rituximab en combinación de metotrexato mostraron una respuesta mejor que los pacientes negativos a ambos.
Los resultados de eficacia en pacientes tratados con rituximab fueron analizados en base al estado de autoanticuerpos previo al comienzo del tratamiento. En la semana 24, los pacientes que eran seropositivos al FR y/o anti-CCP de base tenían una probabilidad significativamente mayor de alcanzar respuesta ACR 20 y 50 comparado con los pacientes seronegativos (p=0,0312 y p=0,0096) (Tabla 18). Estos resultados se repitieron en la Semana 48, cuando la seropositividad a los autoanticuerpos también incrementó de forma significativa la probabilidad de alcanzar ACR70. En la semana 48, los pacientes seropositivos tenían una probabilidad 2-3 veces mayor de alcanzar respuestas ACR comparado con los pacientes seronegativos. Los pacientes seropositivos también tuvieron un descenso significativamente mayor en DAS28-ESR en comparación con los pacientes seronegativos. (Figura 1).
Tabla 18 Resumen de eficacia del estado basal de anticuerpos
| Semana 24 | Semana 48 | |||
| Seropositivo (n = 514) | Seronegativo (n = 106) | Seropositivo (n = 506) | Seronegativo (n = 101) | |
| ACR20 (%) | 62,3* | 50,9 | 71,1* | 51,5 |
| ACR50 (%) | 32,7* | 19,8 | 44,9** | 22,8 |
| ACR70 (%) | 12,1 | 5,7 | 20,9* | 6,9 |
| Respuesta EULAR (%) | 74,8* | 62,9 | 84,3* | 72,3 |
| Diferencia media en la DAS28-ESR | –1,97** | –1,50 | –2,48*** | –1,72 |
Los niveles de significancia se definieron como * p < 0,05, ** p < 0,001, *** p < 0,0001.
Figura 1 Cambio de DAS28-ESR basal según el estado basal de autoanticuerpos
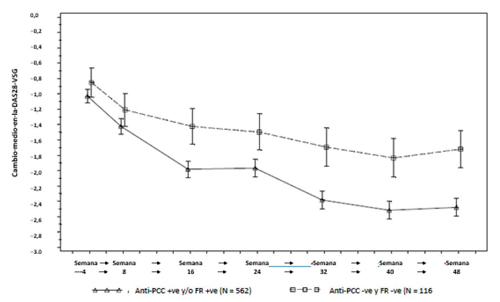
Eficacia a largo plazo con ciclos múltiples de tratamiento
El tratamiento con rituximab en combinación con metotrexato en ciclos múltiples dio lugar a mejoras prolongadas en los signos y síntomas clínicos de la AR, según lo indicado por las respuestas ACR, DAS28-ESR y EULAR, que fueron patentes en todas las poblaciones de pacientes estudiadas (Figura 2). Mejoras sostenidas en la función física según lo indicado por la puntuación del HAQ-DI y la proporción de pacientes que alcanzaron una MPCI en el HAQ-DI.
Figura 2 Respuestas ACR para 4 ciclos de tratamiento (24 semanas después de cada ciclo [dentro de los pacientes, dentro de las visitas]) en pacientes con una respuesta inadecuada a los inhibidores del TNF (n = 146)
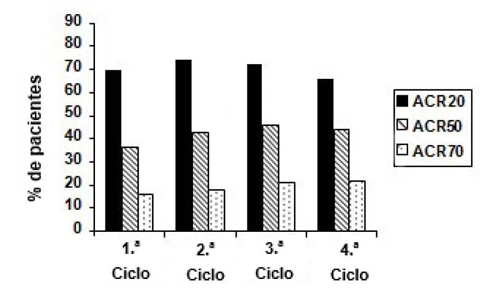
Hallazgos clínicos de laboratorio
En total, 392/3095 (12,7 %) pacientes con artritis reumatoide dieron un resultado positivo de ADA en los análisis clínicos efectuados después del tratamiento con rituximab. La aparición de ADA no se asoció a ningún deterioro clínico ni aumentó el riesgo de reacciones a las perfusiones posteriores en la mayoría de estos pacientes. La presencia de los ADA puede estar asociada con un empeoramiento de las reacciones asociadas a la perfusión o de las reacciones alérgicas después de la segunda perfusión de los ciclos siguientes.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con rituximab en todos los grupos de la población pediátrica con artritis autoinmune. Ver sección 4.2 para consultar la información sobre el uso en la población pediátrica.
Experiencia clínica en granulomatosis con poliangeitis (GPA) y poliangeitis microscópica (PAM)
Inducción de la remisión en adultos
En el estudio 1 de GPA/PAM, un total de 197 pacientes de 15 años o más con GPA (75 %) y PAM (24 %) activas y graves se incluyeron y trataron en un ensayo de no-inferioridad, multicéntrico, doble ciego, aleatorizado con comparador activo y controlado.
Los pacientes fueron aleatorizados en una proporción de 1:1 para recibir ciclofosfamida por vía oral diariamente (2 mg/kg/día) durante 3-6 meses o rituximab (375 mg/m2) una vez por semana durante 4 semanas. Todos los pacientes en el brazo de ciclofosfamida recibieron tratamiento de mantenimiento con azatioprina durante el seguimiento. Los pacientes en ambos brazos recibieron 1.000 mg de metilprednisolona en bolo intravenoso (IV) (u otra dosis equivalente de glucocorticoides) por día, de 1 a 3 días, seguida de prednisona por vía oral (1 mg/kg/día, sin exceder 80 mg/día). La disminución gradual de prednisona debe completarse a los 6 meses desde el inicio del tratamiento del ensayo.
La variable principal fue conseguir la remisión completa a los 6 meses, definida como el Índice de actividad de vasculitis de Birmingham para la granulomatosis de Wegener (BVAS/WG) de 0, y sin tratamiento con glucocorticoides. El margen preespecificado de no inferioridad fue del 20 %. El ensayo demostró la ausencia de inferioridad de rituximab frente a ciclofosfamida para la remisión completa a los 6 meses (Tabla 19).
Se observó eficacia tanto en pacientes con enfermedad recién diagnosticada como en pacientes con una enfermedad recurrente (Tabla 20).
Tabla 19 Porcentaje de pacientes adultos que alcanzaron la remisión completa a los 6 meses (población por intención de tratar*)
| Rituximab (n = 99) | Ciclofosfamida (n = 98) | Tratamiento diferente a (Rituximab-Ciclofosfamida) | |
| Tasa | 63,6 % | 53,1 % | 10,6 % IC del 95,1 %b (–3,2 %, 24,3 %)a |
| – IC = intervalo de confianza. – * Imputación del peor caso a La no inferioridad se demostró ya que el límite inferior (– 3,2%) fue superior al margen de no inferioridad predeterminado (– 20%). b El intervalo de confianza del 95,1 % refleja un alfa adicional de 0,001 para tener en cuenta un análisis intermedio de eficacia. | |||
Tabla 20 Remisión completa a los 6 meses por estado de enfermedad
| Rituximab | Ciclofosfamida | Diferencia (IC del 95 %) | |
| Todos los pacientes Recién diagnosticados Recaídas | n = 99 n = 48 n = 51 | n = 98 n = 48 n = 50 | |
| Remisión completa | |||
| Todos los pacientes | 63,6 % | 53,1 % | 10,6 % (–3,2; 24,3) |
| Recién diagnosticados | 60,4 % | 64,6 % | –4,2 % (–23,6; 15,3) |
| Recaídas | 66,7 % | 42,0 % | 24,7 % (5,8; 43,6) |
La imputación del peor caso se aplica a los pacientes con datos incompletos
Remisión completa a los 12 y 18 meses.
En el grupo de rituximab, el 48% de los pacientes lograron la remisión completa (RC) a los 12 meses, y el 39% de los pacientes lograron una remisión completa a los 18 meses. En los pacientes tratados con ciclofosfamida (seguido de azatioprina para mantener la remisión completa), el 39% de los pacientes lograron una RC a los 12 meses y el 33% de los pacientes lograron una RC a los 18 meses. Desde el mes 12 al mes 18, se observaron 8 recaídas en el grupo de rituximab en comparación con las cuatro en el grupo de ciclofosfamida.
Análisis de laboratorio
Un total de 23/99 (23 %) pacientes tratados con rituximab del ensayo de inducción de la remisión dieron positivo para ADA a los 18 meses. Ninguno de los 99 pacientes tratados con rituximab fue positivo para ADA en el cribado. No hubo una tendencia aparente o un impacto negativo aparente de la presencia de ADA en la seguridad o la eficacia en el ensayo de inducción de la remisión.
Tratamiento de mantenimiento en adultos
Un total de 117 pacientes (88 con GPA, 24 con PAM y 5 con vasculitis limitada al riñón relacionada con ANCA) en remisión fueron aleatorizados para recibir azatioprina (59 pacientes) o rituximab (58 pacientes) en un estudio prospectivo, multicéntrico, controlado y abierto. Los pacientes incluidos tenían entre 21 y 75 años y tenían una enfermedad recién diagnosticada o recidivante en remisión completa después del tratamiento combinado con glucocorticoides y ciclofosfamida. La mayoría de pacientes eran ANCA positivos en el momento del diagnóstico o durante el curso de su enfermedad; tenían una vasculitis necrotizante de vasos pequeños confirmada histológicamente con un fenotipo clínico de GPA o PAM o vasculitis renal limitada asociada a ANCA; o ambos.
La terapia de inducción de remisión incluyó prednisona IV, administrada según el criterio del investigador, precedida en algunos pacientes por metilprednisolona y ciclofosfamida hasta que se logró la remisión después de 4 a 6 meses. En ese momento, y dentro de un máximo de 1 mes después de la última administración de ciclofosfamida, los pacientes fueron asignados aleatoriamente para recibir rituximab (dos perfusiones IV de 500 mg separadas por dos semanas (en el Día 1 y Día 15) seguidas de 500 mg IV cada 6 meses durante 18 meses) o azatioprina (administrada por vía oral a una dosis de 2 mg/kg/día durante 12 meses, luego 1.5 mg/kg/día durante 6 meses y finalmente 1 mg/kg/día durante 4 meses (interrupción del tratamiento después de estos 22 meses)). El tratamiento con prednisona se redujo y luego se mantuvo en una dosis baja (aproximadamente 5 mg por día) durante al menos 18 meses después de la aleatorización. La disminución de la dosis de prednisona y la decisión de suspender el tratamiento con prednisona después del mes 18 se dejaron a decisión del investigador.
Todos los pacientes fueron monitorizados hasta el mes 28 (10 o 6 meses, respectivamente, después de la última perfusión de rituximab o la dosis de azatioprina). Se requirió profilaxis para la neumonía por Pneumocystis jirovecii para todos los pacientes con recuentos de linfocitos T CD4 + menores a 250 por milímetro cúbico.
La medida de resultado primario fue la tasa de recaída grave en el mes 28.
Resultados
En el mes 28, una recaída grave (considerada como la reaparición de datos clínicos o analíticos de vasculitis ([BVAS]> 0) que podía llevar a insuficiencia o daño orgánico o podría ser potencialmente mortal) ocurrió en 3 pacientes (5%) en el grupo de rituximab y 17 pacientes (29%) en el grupo de azatioprina (p = 0,0007). Las recaídas menores (que no ponen en peligro la vida y que no implican daño orgánico mayor) ocurrieron en siete pacientes en el grupo de rituximab (12%) y ocho pacientes en el grupo de azatioprina (14%).
Las curvas de tasas de incidencia acumulativa mostraron que el tiempo hasta la primera recaída grave era más prolongado en los pacientes con rituximab a partir del mes 2 y se mantenía hasta el mes 28 (Figura 3).
Figura 3 Incidencia acumulada en el tiempo de la primera recidiva grave
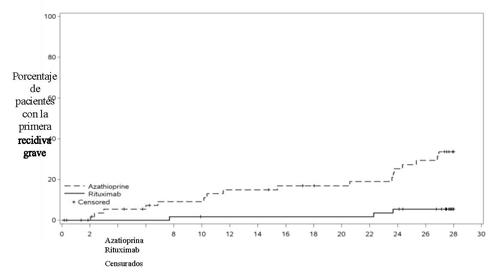
Tiempo de supervivencia (meses)
| Número de sujetos que sufrieron una recidiva grave | |||||||||||||||
| Azatioprina | 0 | 0 | 3 | 3 | 5 | 5 | 8 | 8 | 9 | 9 | 9 | 10 | 13 | 15 | 17 |
| Rituximab | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 3 | 3 | 3 |
| Número de sujetos en riesgo | |||||||||||||||
| Azatioprina | 59 | 56 | 52 | 50 | 47 | 47 | 44 | 44 | 42 | 41 | 40 | 39 | 36 | 34 | 0 |
| Rituximab | 58 | 56 | 56 | 56 | 55 | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 52 | 50 | 0 |
Nota: Los pacientes fueron censurados en el mes 28 si no sufrieron ningún acontecimiento.
Análisis de laboratorio
Un total de 6/34 (18 %) de los pacientes tratados con rituximab del ensayo clínico de terapia de mantenimiento desarrollaron ADA. No hubo una tendencia aparente o un impacto negativo aparente de la presencia de ADA en la seguridad o la eficacia en el ensayo clínico de terapia de mantenimiento.
Población pediátrica
Granulomatosis con poliangeitis (GPA) y poliangeitis microscópica (PAM)
El estudio WA25615 (PePRS) fue un estudio abierto, multicéntrico, de un solo brazo, no controlado en 25 pacientes pediátricos (≥ 2 a < 18 años) con GPA o PAM, activas y graves. La edad media de los pacientes en el estudio fue: 14 años (rango: 6-17 años) y la mayoría de los pacientes (20/25 [80%]) eran mujeres. Un total de 19 pacientes (76 %) tenían GPA y 6 pacientes (24 %) tenían PAM al inicio del estudio. Dieciocho pacientes (72 %) tuvieron una enfermedad recién diagnosticada al ingresar al estudio (13 pacientes con GPA y 5 pacientes con PAM) y 7 pacientes tuvieron una enfermedad recurrente (6 pacientes con GPA y 1 paciente con PAM).
El diseño del estudio consistió en una fase inicial de inducción de remisión de 6 meses y un seguimiento de 18 meses como mínimo hasta un máximo de 54 meses (4,5 años) en total. Los pacientes debían recibir un mínimo de 3 dosis de metilprednisolona por vía intravenosa (30 mg/kg/día, no más de 1 g/día) antes de la primera perfusión de rituximab IV. Si está indicado clínicamente, se pueden administrar dosis diarias adicionales (hasta tres) de metilprednisolona IV. El régimen de inducción de remisión consistió en cuatro perfusiones IV una vez por semana de rituximab a una dosis de 375 mg/m2 de superficie corporal, en los días de estudio 1, 8, 15 y 22 en combinación con prednisolona oral o prednisona a 1 mg/kg/día (máx. 60 mg/día) disminuido a 0,2 mg/kg/día mínimo (máximo 10 mg/día) en el sexto mes. Después de la fase de inducción de remisión, los pacientes podían, bajo criterio del investigador, recibir perfusiones posteriores de rituximab en o después del sexto mes para mantener la remisión PVAS y controlar la actividad de la enfermedad (incluyendo enfermedad progresiva o brote) o para lograr la primera remisión.
Los 25 pacientes completaron las cuatro perfusiones intravenosas una vez por semana para la fase de inducción de remisión de 6 meses. Un total de 24 de 25 pacientes completaron al menos 18 meses de seguimiento.
Los objetivos de este estudio fueron evaluar la seguridad, los parámetros de farmacocinética y la eficacia de rituximab en pacientes con pediátricos GPA y PAM (≥ 2 a < 18 años). Los objetivos de eficacia del estudio fueron exploratorios y se evaluaron principalmente mediante el puntaje de actividad de vasculitis pediátrica (PVAS) (Tabla 21).
Dosis acumulativa de glucocorticoides (IV y oral) al sexto mes
En el Estudio WA25615, veinticuatro de 25 pacientes (96 %) lograron una disminución gradual de los glucocorticoides orales a 0,2 mg/kg/día (o menor o igual a 10 mg/día, lo que fuera menor) en el Mes 6 durante el protocolo de reducción de esteroides orales definido.
Se observó una disminución en la mediana del uso total de glucocorticoides orales desde la semana 1 (mediana = 45 mg de dosis equivalente de prednisona [RIQ: 35 - 60]) hasta el mes 6 (mediana = 7,5 mg [RIQ: 4-10]), que posteriormente se mantuvo en el mes 12 (mediana = 5 mg [RIQ: 2-10]) y en el mes 18 (mediana = 5 mg [RIQ: 1-5]).
Tratamiento de seguimiento
Durante el período total de estudio, los pacientes recibieron entre 4 y 28 perfusiones de rituximab (hasta 4,5 años [53,8 meses]). Los pacientes recibieron hasta 375 mg/m2 x 4 de rituximab, aproximadamente cada 6 meses a criterio del investigador. En total, 17 de 25 pacientes (68 %) recibieron tratamiento adicional con rituximab en o después del mes 6 hasta el cierre común, 14 de estos 17 pacientes recibieron tratamiento adicional con rituximab entre el mes 6 y el mes 18.
Tabla 21 Estudio WA25615 (PePRS) - Remisión PVAS a mes 1, 2, 4, 6, 12 y 18
| Visita de estudio | Número de pacientes respondedores en remisión PVAS* (ratio de respuesta [%]) N = 25 | 95 % ICα |
| Mes 1 | 0 | 0,0 %, 13,7 % |
| Mes 2 | 1 (4,0 %) | 0,1 %, 20,4 % |
| Mes 4 | 5 (20,0 %) | 6,8 %, 40,7 % |
| Mes 6 | 13 (52,0 %) | 31,3 %, 72,2 % |
| Mes 12 | 18 (72,0 %) | 50,6 %, 87,9 % |
| Mes 18 | 18 (72,0 %) | 50,6 %, 87,9 % |
| *PVAS de 0 y nivel de glucocorticoides reducido a 0,2 mg/kg/día (o 10 mg/día, lo que sea menor) en el momento de la evaluación. α Los resultados de eficacia son exploratorios y no se realizaron pruebas estadísticas formales para estas variables El tratamiento con rituximab, (375 mg/m2 x 4 infusiones) hasta el mes 6 fue idéntico para todos los pacientes. El tratamiento de seguimiento posterior al Mes 6 quedó a criterio del investigador. | ||
Evaluaciones de laboratorio
Un total de 4/25 pacientes (16 %) desarrollaron ADA durante todo el período de estudio. Los limitados datos muestran que no se observó una tendencia en las reacciones adversas informadas en pacientes con ADA positivos.
No hubo una tendencia aparente o un impacto negativo de la presencia de ADA en la seguridad o la eficacia en los ensayos clínicos pediátricos de GPA y PAM.
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los estudios con rituximab en población pediátrica < 2 años de edad con GPA o PAM activas graves. Consulte la sección 4.2 para obtener información sobre el uso pediátrico.
Experiencia clínica en pénfigo vulgar
Estudio 1 de PV (Estudio ML22196)
La eficacia y seguridad de rituximab en combinación con glucocorticoides a corto plazo y a dosis bajas (prednisona) fue evaluada en pacientes recién diagnosticados con pénfigo de moderado a grave (74 con pénfigo vulgar [PV] y 16 con pénfigo foliáceo [PF]) en este estudio aleatorizado, abierto, controlado , multicéntrico. Los pacientes tenían edades comprendidas entre los 19 y 79 años y no habían recibido tratamiento previo para pénfigo. En la población con PV, 5 (13%) de los pacientes en el grupo de rituximab y 3 (8%) de los pacientes en el grupo con prednisona a dosis estándar sufrieron enfermedad moderada y 33 (87%) de los pacientes en el grupo de rituximab y 33 (92%) de los pacientes en el grupo de prednisona a dosis estándar sufrieron enfermedad grave según los criterios de gravedad definidos por Harman.
Los pacientes se estratificaron según la gravedad de la enfermedad basal (moderada o grave) y se aleatorizaron 1:1 para recibir rituximab con prednisona a dosis bajas o prednisona a dosis estándar. Los pacientes randomizados en el grupo de rituximab, recibieron una perfusión inicial de 1000 mg en el día 1 del estudio en combinación con 0,5 mg/kg/día de prednisona oral reducida gradualmente durante 3 meses si tenían enfermedad moderada, o 1 mg/kg/día de prednisona oral reducida gradualmente durante 6 meses si tenían enfermedad grave y una segunda perfusión intravenosa de 1000 mg en el día 15 del estudio. Se administraron perfusiones de mantenimiento con rituximab de 500 mg en los meses 12 y 18. Los pacientes aleatorizados en el grupo de prednisona a dosis estándar, recibieron una dosis inicial de 1 mg/kg/día de prednisona oral reducida gradualmente durante 12 meses si tenían enfermedad moderada o 1,5 mg/kg/día de prednisona oral reducida gradualmente durante 18 meses, si tenían enfermedad grave. Los pacientes en el grupo de rituximab que sufrieron una recaída podían recibir una perfusión adicional de rituximab 1000 mg en combinación con una reintroducción o aumento de la dosis de prednisona. Las perfusiones por recaída o de mantenimiento no se administraban antes de las 16 semanas tras la perfusión anterior.
La variable principal del estudio era la remisión completa (epitelización completa y ausencia de lesiones nuevas y/o existentes) en el mes 24 sin el uso de prednisona durante dos o más meses (CR off durante ≥2 meses).
Resultados del Estudio 1 de PV
El estudio mostró resultados estadísticamente significativos de rituximab y dosis bajas de prednisona sobre la dosis estándar de prednisona para lograr CRoff ≥ 2 meses al mes 24 en pacientes con PV (ver Tabla 22).
Tabla 22 Porcentaje de pacientes con PV que alcanzaron la remisión completa sin tratamiento con corticosteroides durante dos meses o más al mes 24 (Población por intención de tratar-PV)
| Rituximab + Prednisona N = 38 | Prednisona N = 36 | Valor pa | IC del 95 %b | |
| Número de pacientes que respondieron al tratamiento (tasa de respuesta [%]) | 34 (89,5 %) | 10 (27,8 %) | < 0,0001 | 61,7 % (38,4; 76,5) |
aEl valor p asociado al test exacto de Fisher con corrección mid-p
bEl intervalo de confianza del 95 % corregido por el intervalo de Newcombe
El número de pacientes en tratamiento con rituximab y prednisona a dosis bajas, sin prednisona o en tratamiento mínimo de prednisona (dosis de 10 mg o menos al día) en comparación con pacientes con dosis estándar de prednisona durante el período de tratamiento de 24 meses muestra el efecto de rituximab como ahorrador de esteroides (Figura 4).
Figura 4: Número de pacientes sin tratamiento o en tratamiento mínimo con corticosteroides (≤ 10 mg/día) en el tiempo
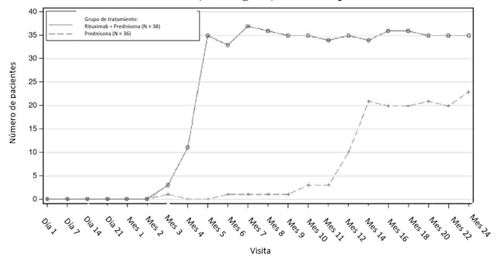
Análisis de laboratorio retrospectivo a posteriori
Un total de 19/34 (56 %) pacientes con PV, que fueron tratados con rituximab, fueron positivos a los anticuerpos ADA a los 18 meses. La relevancia clínica de la formación de ADA en pacientes con PV tratados con rituximab no es clara.
Estudio 2 de PV (Estudio WA29330)
En un estudio aleatorizado, doble ciego, doble simulado, de comparación activa, multicéntrico, se evaluó la eficacia y seguridad de rituximab en comparación con micofenolato mofetilo (MFM) en pacientes con PV de moderado a grave que habían recibido 60-120 mg/día de prednisona oral o equivalente (1,0-1,5 mg/kg/día) en el momento de ingreso en el estudio y se disminuyó gradualmente hasta alcanzar una dosis de 60 u 80 mg/día para el día 1. En pacientes diagnosticados con PV dentro de los 24 meses anteriores y con evidencia de enfermedad de moderada a grave (definida como una Pemphigus Disease Area Index, PDAI calificación de actividad total de ≥ 15).
Ciento treinta y cinco pacientes fueron asignados al azar al tratamiento con rituximab 1000 mg administrados el día 1, día 15, semana 24 y semana 26 o con MFM oral 2 g/día durante 52 semanas en combinación con 60 u 80 mg de prednisona oral con el objetivo de disminuir gradualmente a 0 mg/día de prednisona en la semana 24.
El objetivo primario de eficacia para este estudio fue evaluar en la semana 52, la eficacia de rituximab en comparación con MFM para lograr una remisión completa sostenida definida como lograr la curación de lesiones sin nuevas lesiones activas (es decir, clasificación de actividad PDAI de 0) mientras tomaban 0 mg/día prednisona o equivalente, y mantener esta respuesta durante al menos 16 semanas consecutivas, durante el período de tratamiento de 52 semanas.
Resultados del Estudio 2 de PV
El estudio demostró la superioridad de rituximab sobre MFM en combinación con una disminución gradual de corticosteroides orales alcanzando un nivel de CRoff corticosteroides ≥ 16 semanas en la semana 52 en pacientes con PV (Tabla 23). La mayoría de los pacientes de la población mITT (74 %) fueron recién diagnosticados y el 26 % de los pacientes tenían enfermedad establecida (duración de la enfermedad ≥ 6 meses y habían recibido tratamiento previo para PV).
Tabla 23 Porcentaje de pacientes con PV que lograron una remisión completa mantenida de la terapia con corticosteroides durante 16 semanas o más en la semana 52 (población de intención de tratar modificada)
| Ruxience (N = 62) | MFM (N = 63) | Diferencia (95 % IC) | valor de p | |
| Pacientes con la enfermedad establecida | 6 (42,9 %) | 2 (10,5 %) | ||
| MMF = Micofenolato mofetilo. IC = Intervalo de Confianza. Pacientes recién diagnosticados = duración de la enfermedad <6 meses o ningún tratamiento previo para PV. Pacientes con enfermedad establecida = duración de la enfermedad ≥ 6 meses y habían recibido tratamiento previo para PV. Se usa el test Cochran-Mantel-Haenszel para el valor de p. | ||||
El análisis de todos los parámetros secundarios (incluida la dosis acumulada de corticosteroides orales, el número total de brotes de enfermedades y el cambio en la calidad de vida relacionada con la salud, según lo medido por el Índice de calidad de vida de dermatología) verificó la mejoría estadísticamente significativa de rituximab en comparación con MMF. Las pruebas de las variables secundarias se controlaron por multiplicidad.
Exposición a glucocorticoides
La dosis acumulada de corticosteroides orales fue significativamente menor en pacientes tratados con rituximab. La mediana (min, max) de la dosis acumulada de prednisona en la semana 52 fue de 2775 mg (450, 22180) en el grupo de pacientes con rituximab en comparación con 4005 mg (900, 19920) en el grupo de pacientes con MMF (p = 0,0005).
Brote de enfermedad
El número total de brotes de enfermedad en pacientes tratados con rituximab fue significativamente menor en comparación con MMF (6 frente a 44, p < 0,0001) y hubo menos pacientes que tuvieron al menos un brote de enfermedad (8,1 % frente a 41,3 %).
Evaluaciones de laboratorio
En la semana 52, un total de 20/63 (31,7 %) (19 inducidos por el tratamiento y 1 aumentado por el tratamiento) pacientes con PV tratados con rituximab dieron positivo en ADA. No hubo impacto negativo aparente de la presencia de ADA en la seguridad o eficacia del Estudio 2 de PV.
5.2 - Propiedades farmacocinéticas de RUXIENCE 100 mg Concent. para sol. para perfus.
Linfoma no Hodgkin en adultos
Sobre un análisis farmacocinético poblacional en 298 pacientes con LNH que recibieron perfusiones únicas o múltiples de rituximab bien solo o bien en combinación con terapia CHOP (el intervalo de dosis administradas de rituximab fue de 100 a 500 mg/m2), los parámetros poblacionales típicos de aclaramiento no específico (CL1), aclaramiento específico (CL2) a los que probablemente contribuyeron las células B o la carga tumoral, y el volumen de distribución en el compartimento central (V1) se estimaron en 0,14 l/día, 0,59 l/día y 2,7 l, respectivamente. La mediana de la semivida de eliminación terminal estimada de rituximab fue 22 días (intervalo 6,1 a 52 días). Los recuentos basales de células CD19-positivas y el tamaño de las lesiones tumorales medibles contribuyeron a cierta variabilidad en el CL2 de rituximab en los datos de 161 pacientes que recibieron 375 mg/m2 en forma de una perfusión intravenosa semanal durante 4 semanas. Los pacientes con mayores recuentos de células CD-19 positivas o lesiones tumorales tuvieron un CL2 más alto. Sin embargo, seguía existiendo una gran variabilidad interindividual para el CL2 después de corregirlo según los recuentos de células CD19-positivas y el tamaño de la lesión tumoral. El V1 varió en función del área de la superficie corporal (ASC) y la terapia CHOP. Esta variabilidad en el V1 (27,1 % y 19,0 %), a la que contribuyeron el rango de la variable superficie corporal (1,53 a 2,32 m2) y la terapia CHOP concomitante, respectivamente, fue relativamente pequeña. Edad, sexo y estado funcional de la OMS no tuvieron efecto alguno sobre la farmacocinética de rituximab. Este análisis indica que no es de esperar que el ajuste de la dosis de rituximab en función de cualquiera de las covariables estudiadas conduzca a una reducción significativa en su variabilidad farmacocinética.
La administración mediante perfusión intravenosa de 4 dosis de 375 mg/m2 cada una de rituximab a intervalos semanales, a 203 pacientes con LNH que recibían por primera vez rituximab dio lugar a una Cmax media tras la cuarta perfusión de 486 μg/ml (intervalo 77,5 a 996,6 μg/ml). Se detectó rituximab en el plasma de los pacientes a los 3-6 meses de finalizar el último tratamiento.
Al administrar 8 dosis de 375 mg/m2 cada una de rituximab en perfusión intravenosa a intervalos semanales a 37 pacientes con LNH, la media de la Cmax aumentó con cada perfusión sucesiva, comprendiendo desde una media de 243 μg/ml (intervalo 16 – 582 μg/ml) tras la primera perfusión hasta 550 μg/ml (intervalo 171 – 1177 μg/ml) tras la octava perfusión
El perfil farmacocinético de rituximab cuando se administró en 6 perfusiones de 375 mg/m2 en combinación con 6 ciclos de quimioterapia CHOP fue similar al observado con rituximab sólo.
Pacientes pediátricos con LBDCG/LB/LLA-B madura/LBL
En el ensayo clínico que estudió LBDCG/LB/LLA-B madura/LBL en pacientes pediátricos, se estudiaron los parámetros farmacocinéticos (FC) en un subconjunto de 35 pacientes de 3 años en adelante. Los parámetros FC fueron comparables entre los dos grupos de edad (≥ 3 a < 12 años vs. ≥ 12 a < 18 años). Después de dos perfusiones de rituximab IV de 375 mg/m2 en cada uno de los dos ciclos de inducción (ciclo 1 y 2) seguido de una perfusión de rituximab IV de 375 mg/m2 en cada uno de los ciclos de consolidación (ciclo 3 y 4) la concentración máxima fue mayor después de la cuarta perfusión (ciclo 2) con una media geométrica de 347 μg/ml seguido de concentraciones máximas medias geométricas más bajas a partir de entonces (Ciclo 4: 247 μg/ml). Con este régimen de dosis, los niveles mínimos se mantuvieron (medias geométricas: 41,8 μg/ml (dosis previa al ciclo 2; después de 1 ciclo), 67,7 μg/ml (dosis previa al ciclo 3, después de 2 ciclos) y 58,5 μg/ml (dosis previa al ciclo 4, después de 3 ciclos). La vida media de eliminación en pacientes pediátricos de 3 años y mayores fue de 26 días.
Las características farmacocinéticas de rituximab en pacientes pediátricos con LBDCG/LB/LLA-B madura/LBL fueron similares a las observadas en pacientes adultos con LNH.
No hay datos farmacocinéticos disponibles en el grupo de edad de ≥ 6 meses a < 3 años, sin embargo, la farmacocinética de la población prevista respalda la exposición sistémica comparable (AUC, Ctrough) en este grupo de edad en comparación con ≥ 3 años (Tabla 24). El tamaño del tumor basal más pequeño se relaciona con una exposición más alta debido a un aclaramiento dependiente del tiempo menor, sin embargo, las exposiciones sistémicas impactadas por diferentes tamaños de tumor permanecen en el rango de exposición que fue eficaz y tenía un perfil de seguridad aceptable.
Tabla 24 Parámetros farmacocinéticos previstos siguiendo el régimen de dosificación derituximab en pacientes pediátricos con LBDCG/LB/LLA-B madura/LBL
| Grupo de edad | ≥ 6 meses a < 3 años | ≥ 3 a < 12 años | ≥ 12 a< 18 años |
| Ctrough (μg/ml) | 47,5 (0,01-179) | 51,4 (0,00-182) | 44,1 (0,00-149) |
| AUC1-4 ciclos (μg*día/ml) | 13501 (278-31070) | 11609 (135-31157) | 11467 (110-27066) |
Los resultados se presentan como la mediana (min – max); Ctrough es pre-dosis Ciclo 4.
Leucemia linfocítica crónica
Rituximab se administró como perfusión intravenosa en el primer ciclo con una dosis de 375 mg/m2 aumentando hasta 500 mg/m2 cada ciclo, durante 5 ciclos en combinación con fludarabina y ciclofosfamida en pacientes con LLC. La media de la Cmax (N=15) fue 408 μg/ml (rango, 97 – 764 μg/ml) después de la quinta perfusión de 500 mg/ m2. y la media de la semivida fue 32 días (rango, 14-62 días).
Artritis reumatoide
Después de administrar dos dosis de 1000 mg de rituximab como perfusión intravenosa con un intervalo de dos semanas, la media de la semivida de eliminación terminal fue de 20,8 días (rango 8,58 a 35,9 días); el aclaramiento sistémico medio 0,23 l/día (rango 0,091 a 0,67 l/día); y el volumen de distribución medio en el estado estacionario 4,6 l (rango: 1,7 a 7,51 l). El análisis farmacocinético poblacional de esos mismos datos dio valores parecidos del aclaramiento sistémico y la semivida (0,26 l/día y 20,4 días, respectivamente). El análisis farmacocinético poblacional reveló que la superficie corporal y el género eran las covariables más importantes que justificaban la variabilidad interindividual de los parámetros farmacocinéticos. Después de ajustar por la superficie corporal, los varones mostraron un volumen de distribución mayor y un aclaramiento más rápido que las mujeres. Las diferencias de género en la farmacocinética no se consideran clínicamente relevantes y no exigen ningún ajuste posológico. No se dispone de datos farmacocinéticos en pacientes con insuficiencia hepática o renal.
La farmacocinética de rituximab fue evaluada en 4 ensayos, tras dos dosis intravenosas (IV) de 500 mg y 1000 mg en los días 1 y 15. En todos estos ensayos, la farmacocinética de rituximab fue dosis dependiente dentro del limitado rango de dosis estudiado. La media de Cmax en suero de rituximab después de la primera perfusión osciló de 157 a 171 μg/ml para la dosis de 2 x 500 mg y de 298 a 341 μg/ml para la dosis de 2 x 1000 mg. Después de la segunda perfusión, la media de Cmax osciló de183 a 198 μg/ml para la dosis de 2 x 500 mg y entre 355 a 404 μg/ml para la dosis de 2 x 1000 mg. La semivida de eliminación terminal promedio osciló de 15 a 16 días para el grupo de dosis de 2 x 500 mg y de 17 a 21 días para el grupo de dosis de 2 x 1000 mg. La media de la Cmax fue del 16 al 19 % mayor después de la segunda perfusión comparándolo con la primera perfusión para ambas dosis.
La farmacocinética de rituximab fue evaluada después de dos dosis IV de 500 mg y 1000 mg en el segundo ciclo de retratamiento. La media de Cmax en suero de rituximab después de la primera perfusión fue de 170 a 175 μg/ml para la dosis de 2 x 500 mg y de 317 a 370 μg/ml para la dosis de 2 x 1000 mg. Después de la segunda perfusión la Cmax fue de 207 μg/ml para la dosis de 2 x 500 mg y de 377 a 386 μg/ml para la dosis de 2 x 1000 mg. La semivida de eliminación media terminal después de la segunda perfusión, tras el segundo ciclo fue de 19 días para el grupo de dosis de 2 x 500 mg y de 21 a 22 días para el grupo de dosis de 2 x 1000 mg. Los parámetros farmacocinéticos de rituximab fueron comparables entre los dos ciclos de tratamiento.
Los parámetros farmacocinéticos (PK) en la población con una respuesta inadecuada a los inhibidores de TNF, que recibió la misma posología (2 x 1000 mg, IV con un intervalo de 2 semanas), se asemejaron: media de la Cmax plasmática 369 μg/ml y de la semivida de eliminación terminal de 19,2 días.
Granulomatosis con poliangeitis (GPA) y poliangeitis microscópica (PAM)
Población adulta
En base al análisis farmacocinético poblacional de los datos en los 97 pacientes con granulomatosis con poliangeitis y poliangeitis microscópica que recibieron 375 mg/m2 de rituximab una vez a la semana por cuatro dosis, la mediana de la semivida de eliminación terminal fue de 23 días (intervalo, 9 a 49 días). El aclaramiento medio y el volumen de distribución de rituximab fueron 0,313 l/día (intervalo, 0,116 a 0,726 l/día) y 4,50 l (intervalo 2,25 a 7,39 l) respectivamente. La concentración máxima durante los primeros 180 días (Cmax), la concentración mínima en el día 180 (C180) y el área acumulada bajo la curva durante 180 días (AUC180) fueron (mediana [rango]) 372,6 (252,3-533,5) μg/ml, 2,1 (0-29,3) μg/ml y 10302 (3653-21874) μg/ml*días, respectivamente. Los parámetros farmacocinéticos de rituximab en pacientes adultos con GPA y PAM parecen similares a los observados en pacientes con artritis reumatoide.
Población pediátrica
Según el análisis farmacocinético de la población de 25 niños (6-17 años) con GPA y PAM que recibieron 375 mg/m2 de rituximab una vez por semana durante cuatro dosis, la vida media de eliminación terminal estimada fue de 22 días (rango, 11 a 42 días). El aclaramiento medio y el volumen de distribución de rituximab fueron 0,221 l/día (rango, 0,0996 a 0,381 l/día) y 2,27 l (rango 1,43 a 3,17 l) respectivamente. La concentración máxima durante los primeros 180 días (Cmax), la concentración mínima en el día 180 (C180) y el área acumulada bajo la curva durante 180 días (AUC180) fueron (mediana [rango]) 382,8 (270,6-513,6) μg/ml, 0,9 (0-17,7) μg/ml y 9787 (4838-20446) μg/ml*día, respectivamente. Los parámetros FC de rituximab en pacientes pediátricos con GPA o PAM fueron similares a aquellos en adultos con GPA o PAM, una vez que se tiene en cuenta el efecto del área de superficie corporal en los parámetros de aclaramiento y volumen de distribución.
Pénfigo vulgar
Los parámetros farmacocinéticos en pacientes adultos con PV que reciben rituximab 1000 mg en los días 1, 15, 168 y 182 se resumen en la Tabla 25.
Tabla 25 Farmacocinética poblacional en pacientes adultos con PV del Estudio 2 de PV
| Parámetro | Ciclo de perfusión | |
| 1er ciclo de 1000 mg Día 1 y Día 15 N = 67 | 2° ciclo de 1000 mg Día 168 y Día 182 N = 67 | |
| Mediana (Rango) | 21,0 (9,3-36,2) | 26,5 (16,4-42,8) |
| Media (Rango) | 391 (159-1510) | 247 (128-454) |
| Media (Rango) | 3,52 (2,48-5,22) | 3,52 (2,48-5,22) |
Después de las dos primeras administraciones de rituximab (en los días 1 y 15, correspondientes al ciclo 1), los parámetros farmacocinéticos de rituximab en pacientes con PV fueron similares a los de pacientes con GPA/PAM y pacientes con AR. Tras las dos últimas administraciones (en los días 168 y 182, correspondientes al ciclo 2), el aclaramiento de rituximab disminuyó mientras que el volumen central de distribución se mantuvo sin cambios.
5.3 - Datos preclínicos sobre seguridad de RUXIENCE 100 mg Concent. para sol. para perfus.
Rituximab ha demostrado ser altamente específico para el antígeno CD20 de las células B. Durante estudios de toxicidad en monos cinomolgos no se observó ningún otro efecto que la disminución farmacológica de los células B en sangre periférica y tejido linfático, previsible por el mecanismo farmacológico.
Se han realizado estudios de toxicidad del desarrollo en monos cinomolgos a dosis de hasta 100 mg/kg (tratamiento en los días 20-50 de la gestación) y no se apreciaron indicios de toxicidad fetal debido al rituximab. Sin embargo, se observó una disminución farmacológica dependiente de la dosis de células B en los órganos linfáticos de los fetos, que persistió después del nacimiento y estuvo acompañado por una disminución en el nivel de IgG en los animales recién nacidos afectados. El recuento de células B volvió a la normalidad en estos animales en los 6 meses siguientes al nacimiento y no afectó a la reacción a la vacunación.
No se han realizado pruebas estándar para investigar la mutagenicidad, ya que dichas pruebas no son relevantes para esta molécula. No se han realizado estudios a largo plazo en animales para establecer el potencial carcinogénico de rituximab.
No se han realizado estudios específicos para determinar los efectos de rituximab sobre la fertilidad. En estudios generales de toxicidad en monos cinomolgos no se observó efecto dañino sobre los órganos reproductivos en machos ni hembras.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de RUXIENCE 100 mg Concent. para sol. para perfus.
L-histidina
Clorhidrato de L-histidina monohidrato
Edetato disódico
Polisorbato 80 (E433)
Sacarosa
Agua para preparaciones inyectables
6.2 - Incompatibilidades de RUXIENCE 100 mg Concent. para sol. para perfus.
No se han observado incompatibilidades entre Ruxience y las bolsas de policloruro de vinilo o polietileno o los equipos de perfusión.
6.3 - Período de validez de RUXIENCE 100 mg Concent. para sol. para perfus.
Vial sin abrir
24 meses
Medicamento diluido
- Después de una dilución aséptica en solución de cloruro de sodio.
La solución de perfusión de Ruxience preparada en solución de cloruro de sodio al 0,9 % es física y químicamente estable durante 35 días a 2 °C-8 °C y durante 24 horas adicionales a ≤ 30 °C.
- Después de una dilución aséptica en solución de D-glucosa
La solución de perfusión de Ruxience preparada en solución de D-glucosa al 5 % es física y químicamente estable durante 24 horas a 2 °C-8 °C y durante 24 horas adicionales a ≤ 30 °C.
Desde un punto de vista microbiológico, la solución de perfusión preparada se debe utilizar inmediatamente. Si no se usa inmediatamente, el tiempo y condiciones de almacenamiento en uso antes de su uso son responsabilidad del usuario y normalmente no durará más de 24 horas a 2 °C-8 °C, a menos que la dilución haya tenido lugar en condiciones asépticas controladas y validadas.
6.4 - Precauciones especiales de conservación de RUXIENCE 100 mg Concent. para sol. para perfus.
Conservar en nevera (entre 2 °C y 8 °C). Conservar el envase en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 - Naturaleza y contenido del recipiente de RUXIENCE 100 mg Concent. para sol. para perfus.
Ruxience 100 mg concentrado para solución para perfusión
Viales de vidrio de tipo I transparentes con tapón de goma de clorobutilo con 100 mg de rituximab en 10 ml.
Envase de 1 vial.
Ruxience 500 mg concentrado para solución para perfusión
Viales de vidrio de tipo I transparentes con tapón de goma de clorobutilo con 500 mg de rituximab en 50 ml.
Envase de 1 vial.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de RUXIENCE 100 mg Concent. para sol. para perfus.
Ruxience se proporciona en viales estériles, sin conservantes y no pirogénicos de uso único.
Utilice una aguja y una jeringa estéril para preparar Ruxience. Extraer asépticamente la cantidad necesaria de Ruxience y diluir a una concentración calculada de 1 a 4 mg/ml de rituximab en una bolsa de perfusión que contenga una solución inyectable estéril libre de pirógenos de cloruro de sodio 9 mg/ml (0,9 %) o 5 % de D-glucosa en agua. Para mezclar la solución, invertir suavemente la bolsa para evitar la formación de espuma. Se debe tener cuidado para garantizar la esterilidad de las soluciones preparadas. Dado que el medicamento no contiene conservantes antimicrobianos ni agentes bacteriostáticos, se debe utilizar una técnica aséptica. Los medicamentos parenterales se deben inspeccionar visualmente para detectar partículas y cambio de color antes de la administración.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Bruxelles
Bélgica
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Ruxience 100 mg concentrado para solución para perfusión
EU/1/20/1431/001
Ruxience 500 mg concentrado para solución para perfusión
EU/1/20/1431/002
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 01 Abril 2020
10. - FECHA DE LA REVISIÓN DEL TEXTO
08/2023.
La información detallada sobre este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
PRESENTACIONES Y PVP (IVA)*:
Ruxience 100 mg concentrado para solución para perfusión, 1 vial:
PVL: 210,45€ ; PVP: 261,36€; PVP IVA: 271,81€.
Ruxience 500 mg concentrado para solución para perfusión, 1 vial:
PVL: 1049,35€; PVP: 1105,26€; PVP IVA: 1149,47€.
CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN:
Medicamento sujeto a prescripción médica. Uso hospitalario.
CONDICIONES DE LA PRESTACIÓN FARMACÉUTICA:
Financiado por el Sistema Nacional de Salud sin aportación.
Para información adicional, por favor, contacte con el Centro de Información Médico-Farmacéutica llamando al + 34 914909900 o consulte nuestra página web www.pfizer.es.

