BOSULIF 400 MG COMPRIMIDOS RECUBIERTOS CON PELICULA

| ATC: Bosutinib |
| PA: Bosutinib monohidrato |
Envases
- Env. con 28
- H: Medicamento de uso hospitalario
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- Fi: Medicamento financiado sólo para determinadas indicaciones
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 721958
- EAN13: 8470007219583
- Conservar en frío: No
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
BOSULIF 400 mg Comp. recub. con película
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Bosulif 100 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 100 mg de bosutinib (como monohidrato).
Bosulif 400 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 400 mg de bosutinib (como monohidrato).
Bosulif 500 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene 500 mg de bosutinib (como monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Comprimido recubierto con película.
Bosulif 100 mg comprimidos recubiertos con película
Comprimido recubierto con película de color amarillo y con forma ovalada biconvexa (ancho: 5,6 mm; longitud: 10,7 mm), marcado con “Pfizer” en una cara y con “100” en la otra.
Bosulif 400 mg comprimidos recubiertos con película
Comprimido recubierto con película de color naranja y con forma ovalada biconvexa (ancho: 8,8 mm; longitud: 16,9 mm), marcado con “Pfizer” en una cara y con “400” en la otra.
Bosulif 500 mg comprimidos recubiertos con película
Comprimido recubierto con película de color rojo y con forma ovalada biconvexa (ancho: 9,5 mm; longitud: 18,3 mm), marcado con “Pfizer” en una cara y con “500” en la otra.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de BOSULIF 400 mg Comp. recub. con película
Bosulif está indicado para el tratamiento de pacientes adultos con:
- Leucemia mieloide crónica con cromosoma Philadelphia positivo (LMC Ph+) en fase crónica (FC) recién diagnosticada.
- LMC Ph+ en FC, fase acelerada (FA) o fase blástica (FB) tratados previamente con uno o más inhibidores de la tirosina quinasa [TKI(s), por sus siglas en inglés] y para quienes imatinib, nilotinib y dasatinib no se consideran opciones adecuadas de tratamiento.
4.2 - Posología y administración de BOSULIF 400 mg Comp. recub. con película
4.3 - Contraindicaciones de BOSULIF 400 mg Comp. recub. con película
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Insuficiencia hepática (ver las secciones 5.1 y 5.2).
4.4 - Advertencias y Precauciones de BOSULIF 400 mg Comp. recub. con película
Alteraciones de la función hepática
El tratamiento con bosutinib se asocia con aumentos en las transaminasas séricas (alanina aminotransferasa [ALT], aspartato aminotransferasa [AST]).
Dichos aumentos se produjeron generalmente al comienzo del tratamiento (de los pacientes que experimentaron un aumento de transaminasas de cualquier grado, > 80% de dichos pacientes experimentaron el primer evento dentro de los 3 primeros meses). Los pacientes en tratamiento con bosutinib deben hacerse pruebas de la función hepática antes del inicio del tratamiento y mensualmente durante los 3 primeros meses de tratamiento, y según indicación clínica.
Los pacientes con aumento de las transaminasas han de manejarse mediante interrupción temporal del tratamiento con bosutinib (considerando que tras recuperación a Grado 1 o al nivel basal puede hacerse una reducción de la dosis), o mediante suspensión definitiva de bosutinib. Los aumentos de transaminasas, especialmente si tienen lugar con incrementos concomitantes de bilirrubina, pueden constituir una señal temprana de lesión hepática inducida por el medicamento, por lo que estos pacientes han de ser manejados adecuadamente (ver las secciones 4.2 y 4.8).
Diarrea y vómitos
El tratamiento con bosutinib se asocia con diarrea y vómitos, por lo que pacientes con alteraciones gastrointestinales clínicamente significativas recientes o en curso deberían utilizar este medicamento con precaución y únicamente tras una cuidadosa evaluación del riesgo-beneficio, ya que pacientes de estas características fueron excluidos de los ensayos clínicos. A los pacientes con diarrea y vómitos se les debe proporcionar el tratamiento habitual, que incluye un medicamento antidiarreico o antiemético y/o la reposición de líquidos. Además, la diarrea y los vómitos pueden controlarse interrumpiendo temporalmente el tratamiento con bosutinib, reduciendo la dosis o con la retirada definitiva de bosutinib (ver las secciones 4.2 y 4.8). El agente antiemético, domperidona, tiene el potencial de prolongar el intervalo QT (QTc) y de inducir arritmias - “torsade de pointes”; por ello, debe evitarse coadministrar domperidona. Solo debe usarse en caso de que el resto de medicamentos no resulten eficaces. En situaciones de este tipo es obligatorio realizar una evaluación individualizada de la relación riesgo/beneficio, y debe vigilarse la posible aparición de una prolongación del intervalo QTc en los pacientes.
Mielosupresión
El tratamiento con bosutinib se asocia con mielosupresión, definida como anemia, neutropenia y trombocitopenia. Deben realizarse recuentos sanguíneos completos semanalmente durante el primer mes, y a partir de ahí, mensualmente, o según indicación clínica. La mielosupresión debe o puede controlarse interrumpiendo temporalmente el tratamiento con bosutinib, reduciendo la dosis o con la retirada definitiva de bosutinib (ver las secciones 4.2 y 4.8).
Retención de líquidos
El tratamiento con bosutinib puede asociarse a una retención de líquidos, incluyendo derrame pericárdico, derrame pleural, edema pulmonar y/o edema periférico. Los pacientes se deben monitorizar y controlar utilizando el tratamiento habitual. Además, la retención de líquidos puede controlarse interrumpiendo temporalmente el tratamiento con bosutinib, reduciendo la dosis o con la retirada definitiva de bosutinib (ver las secciones 4.2 y 4.8).
Lipasa sérica
Se ha observado aumento de la lipasa sérica. Se recomienda proceder con precaución en los pacientes con antecedentes de pancreatitis. En caso de que los aumentos de la lipasa se presenten acompañados de síntomas abdominales, debe interrumpirse el tratamiento con bosutinib y adoptar las medidas diagnósticas que se consideren adecuadas para excluir pancreatitis (ver sección 4.2).
Infecciones
Bosutinib puede predisponer a los pacientes a infecciones bacterianas, fúngicas, víricas o protozoarias.
Potencial proarrítmico
Se ha observado prolongación de la repolarización cardiaca ventricular (intervalo QTc) según la lectura automática realizada por el electrocardiógrafo, sin arritmia concomitante. Bosutinib debe administrarse con precaución a los pacientes con antecedentes o predisposición a la prolongación del intervalo QTc, que tengan una cardiopatía no controlada o significativa incluyendo infarto de miocardio reciente, insuficiencia cardiaca congestiva, angina inestable o bradicardia clínicamente significativa, o que estén recibiendo medicamentos con un efecto conocido de prolongación del QTc (por ejemplo, antiarrítmicos u otros productos que puedan prolongar el QTc [sección 4.5]). La presencia de hipopotasemia e hipomagnesemia puede potenciar este efecto.
Resulta aconsejable realizar una monitorización para ver si aparece algún efecto en el QTc, y se recomienda realizar un electrocardiograma (ECG) basal antes de iniciar el tratamiento con bosutinib, y cuando esté clínicamente indicado. Antes de administrar bosutinib se debe corregir la hipopotasemia o la hipomagnesemia, y se han de monitorizar periódicamente durante el tratamiento.
Insuficiencia renal
El tratamiento con bosutinib puede provocar una disminución clínicamente significativa de la función renal en pacientes con LMC. Se ha observado una disminución a lo largo del tiempo de la tasa de filtración glomerular estimada (TFGe) en pacientes que recibieron tratamiento con bosutinib en ensayos clínicos. En pacientes con LMC en FC recién diagnosticada tratados con 400 mg, la mediana de disminución de la TFGe en relación con el valor basal fue de 11,1 ml/min/1,73 m2 al año y 14,1 ml/min/1,73 m2 a los 5 años para pacientes en tratamiento. Los pacientes con LMC no tratados previamente y que se trataron con 500 mg mostraron una mediana de disminución de TFGe de 9,2 ml/min/1,73 m2 al año, 12,0 ml/min/1,73 m2 a los 5 años y 16,6 ml/min/1,73 m2 a los 10 años para pacientes en tratamiento. En pacientes con LMC en FC y en fase avanzada tratada previamente con 500 mg, la mediana de disminución de la TFGe fue de 7,6 ml/min/1,73 m2 al año, 12,3 ml/min/1,73 m2 a los 5 años y 15,9 ml/min/1,73 m2 a los 10 años para pacientes en tratamiento. En pacientes con LMC Ph+ tratada previamente con 1 o más TKI tratados con 500 mg, la mediana de disminución de la TFGe con respecto al valor basal fue de 9,2 ml/min/1,73 m2 al año y de 14,5 ml/min/1,73 m2 a los 4 años para los pacientes en tratamiento.
Es importante evaluar la función renal antes de iniciar el tratamiento y supervisarla minuciosamente durante el tratamiento con bosutinib, prestando especial atención en el caso de aquellos pacientes que tienen una insuficiencia renal preexistente o factores de riesgo de disfunción renal, incluyendo el uso concomitante de medicamentos que puedan provocar nefrotoxicidad, como diuréticos, inhibidores de la enzima convertidora de angiotensina (ECA), bloqueadores de los receptores de la angiotensina y medicamentos antiinflamatorios no esteroideos (AINEs).
En un estudio sobre la insuficiencia renal, las exposiciones a bosutinib se aumentaron en pacientes con la función renal moderada y gravemente afectada. Se recomienda reducir la dosis en pacientes con insuficiencia renal moderada o grave (ver las secciones 4.2 y 5.2).
En los estudios de LMC se excluyó a los pacientes con una creatinina sérica > 1,5 veces el LSN. De acuerdo a un análisis de las características farmacocinéticas, se observó que, al inicio del tratamiento, durante los estudios se produjo un aumento de la exposición (AUC) en los pacientes con una insuficiencia renal moderada y grave (ver las secciones 4.2 y 5.2).
Los datos clínicos son muy limitados (n = 3) para pacientes con LMC e insuficiencia renal moderada que reciben una dosis aumentada de 600 mg de bosutinib.
Raza asiática
Según los análisis farmacocinéticos poblacionales, la población asiática tuvo un aclaramiento menor, lo que aumentó la exposición. Por lo tanto, estos pacientes se deben someter a una estrecha monitorización para detectar reacciones adversas, especialmente en caso de aumento de la dosis.
Reacciones cutáneas graves
Bosutinib puede provocar reacciones cutáneas graves, tales como el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica. El tratamiento con bosutinib se debe interrumpir de forma permanente en los pacientes que experimenten una reacción cutánea grave durante el tratamiento.
Síndrome de lisis tumoral
Debido a la posible aparición del síndrome de lisis tumoral (SLT), se recomienda la corrección de la deshidratación clínicamente significativa y el tratamiento de los niveles altos de ácido úrico antes de iniciar el tratamiento con bosutinib (ver sección 4.8).
Reactivación del virus de la hepatitis B
Se ha producido reactivación de la hepatitis B en pacientes que son portadores crónicos de este virus después de que los pacientes hayan recibido TKIs BCR-ABL. En algunos casos se produjo insuficiencia hepática aguda o hepatitis fulminante que dio lugar a un trasplante de hígado o a un desenlace mortal.
Los pacientes se deben someter a pruebas para detectar la infección por VHB antes de comenzar el tratamiento con bosutinib. Se debe consultar a expertos en enfermedades hepáticas y en el tratamiento de la hepatitis B antes de comenzar el tratamiento en pacientes con una serología positiva para hepatitis B (incluyendo a los pacientes con enfermedad activa) y pacientes que den un resultado positivo en una prueba de infección por VHB durante el tratamiento. Los portadores del VHB que necesiten tratamiento con bosutinib se deben someter a una estrecha monitorización para detectar signos y síntomas de infección activa por VHB a lo largo de todo el tratamiento y durante varios meses después de finalizar el tratamiento (ver sección 4.8).
Fotosensibilidad
Se debe evitar o minimizar la exposición a la luz solar directa o a la radiación ultravioleta (UV) debido al riesgo de fotosensibilidad asociado al tratamiento con bosutinib. Se debe recomendar a los pacientes que utilicen medidas como ropa protectora y crema de protección solar con factor de protección solar (FPS) alto.
Inhibidores del citocromo P-450 (CYP)3A
Debe evitarse el uso concomitante de bosutinib con inhibidores potentes o moderados del CYP3A, ya que se produciría un aumento de la concentración plasmática de bosutinib (ver sección 4.5).
Se recomienda seleccionar, si es posible, un medicamento concomitante alternativo cuyo potencial de inhibición del CYP3A sea nulo o mínimo.
Si durante el tratamiento con bosutinib resulta necesario administrar un inhibidor potente o moderado del CYP3A, hay que plantearse interrumpir el tratamiento con bosutinib o reducir la dosis de bosutinib.
Inductores del CYP3A
Debe evitarse el uso concomitante de bosutinib con inductores potentes o moderados del CYP3A, ya que se produciría una disminución de la concentración plasmática de bosutinib (ver sección 4.5).
Efecto de los alimentos
Debe evitarse el consumo de productos que contengan pomelo, incluyendo el zumo de pomelo, así como de otros alimentos con un efecto conocido de inhibición del CYP3A (ver sección 4.5).
Contenido en sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido de 100 mg, 400 mg o 500 mg. Se puede informar a los pacientes con dietas bajas en sodio que este medicamento está esencialmente “exento de sodio”.
4.5 - Interacciones con otros medicamentos de BOSULIF 400 mg Comp. recub. con película
Efectos de otros medicamentos sobre bosutinib
Inhibidores del CYP3A
Debe evitarse el uso concomitante de bosutinib con inhibidores potentes del CYP3A (entre otros, itraconazol, ketoconazol, posaconazol, voriconazol, claritromicina, telitromicina, nefazodona, mibefradilo, indinavir, lopinavir/ritonavir, nelfinavir, ritonavir, saquinavir, boceprevir, telaprevir o productos con pomelo, incluido el zumo de pomelo) o con inhibidores moderados del CYP3A (entre otros, fluconazol, ciprofloxacino, eritromicina, diltiazem, verapamilo, amprenavir, atazanavir, darunavir/ritonavir, fosamprenavir, aprepitant, crizotinib o imatinib), ya que se produciría un aumento de la concentración plasmática de bosutinib.
Si se utilizan inhibidores débiles del CYP3A de forma concomitante con bosutinib, debe procederse con precaución.
Se recomienda seleccionar, si es posible, un medicamento concomitante alternativo cuyo potencial de inhibición del enzima CYP3A sea nulo o mínimo.
Si durante el tratamiento con bosutinib, resulta necesario administrar un inhibidor potente o moderado del CYP3A, hay que plantearse interrumpir el tratamiento con bosutinib o reducir la dosis de bosutinib.
En un estudio realizado con 24 sujetos sanos, a quienes se administraron 5 dosis diarias de 400 mg de ketoconazol (un inhibidor potente del CYP3A) junto con una dosis única de 100 mg de bosutinib en ayunas, ketoconazol multiplicó por 5,2 la Cmax de bosutinib, y por 8,6 el AUC de bosutinib en plasma, en comparación con la administración de bosutinib sin ningún otro medicamento.
En un estudio con 20 sujetos sanos, a los que se administró una dosis única de 125 mg de aprepitant (un inhibidor moderado del CYP3A) junto con una dosis única de 500 mg de bosutinib después de recibir alimentos, el aprepitant aumentó 1,5 veces la Cmax de bosutinib y 2,0 veces el AUC de bosutinib en el plasma, en comparación con la administración de bosutinib solo.
Inductores del CYP3A
Debe evitarse el uso concomitante de bosutinib con inductores potentes del CYP3A (entre otros, carbamacepina, fenitoína, rifampicina o hierba de San Juan) o con inductores moderados del CYP3A (entre otros, bosentán, efavirenz, etravirina, modafinilo o nafcilina), ya que se produciría una disminución de la concentración plasmática de bosutinib.
Teniendo en cuenta la gran reducción de la exposición al bosutinib que se produjo cuando se administró bosutinib de forma concomitante con rifampicina, resulta improbable que el aumento de la dosis de bosutinib al administrarlo de forma concomitante con inductores potentes o moderados del CYP3A sea suficiente para compensar la pérdida de exposición.
Si se utilizan inductores débiles del CYP3A de forma concomitante con bosutinib, debe procederse con precaución.
Tras la administración concomitante de una dosis única de bosutinib con 6 dosis diarias de 600 mg de rifampicina en 24 sujetos sanos que habían recibido alimentos, la exposición al bosutinib (Cmax y AUC en plasma) se redujo en un 14% y un 6% respectivamente, respecto a los valores de bosutinib 500 mg administrado sin ningún otro medicamento.
Inhibidores de la bomba de protones (IBP)
Debe procederse con precaución en caso de que bosutinib se administre de forma concomitante con IBP. Siempre que sea posible, debe considerarse el uso de antiácidos de acción corta como alternativa a los IBP, distanciando los momentos de administración de bosutinib y de los antiácidos (es decir, toma de bosutinib por la mañana y de los antiácidos por la noche). Bosutinib muestra solubilidad acuosa in vitro dependiente de pH. En un estudio que se llevó a cabo cuando se administró una dosis única, por vía oral, de bosutinib (400 mg) junto con varias dosis por vía oral de lansoprazol (60 mg) en 24 sujetos sanos en ayunas, la Cmax y el AUC de bosutinib se redujeron en un 54% y un 74%, respectivamente, respecto a los valores de bosutinib (400 mg) administrado sin ningún otro medicamento.
Efectos de bosutinib sobre otros medicamentos
En un estudio con 27 sujetos sanos, a los que se administró una dosis única de 500 mg de bosutinib con una dosis única de 150 mg de mesilato de etexilato de dabigatrán (un sustrato de la P-glicoproteína [P-gp]) después de recibir alimentos, bosutinib no aumentó la Cmax ni el AUC de dabigatrán en el plasma, en comparación con la administración de mesilato de etexilato de dabigatrán solo. Los resultados del estudio indican que bosutinib no presenta efectos inhibitorios clínicamente importantes de la P-gp.
Otro estudio in vitro indica que es improbable que aparezcan interacciones medicamentosas a la dosis terapéutica, como consecuencia de la inducción por parte de bosutinib sobre el metabolismo de medicamentos que sean sustratos del CYP1A2, CYP2B6, CYP2C9, CYP2C19 o CYP3A4.
Estudios in vitro muestran que es improbable que aparezcan interacciones medicamentosas a la dosis terapéutica, como consecuencia de la inhibición por parte de bosutinib sobre el metabolismo de medicamentos que sean sustratos del CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP3A4/5.
Los estudios in vitro indican que bosutinib tiene un bajo potencial para inhibir la proteína de resistencia en el cáncer de mama (BCRP por sus siglas en inglés, sistémicamente), al polipéptido transportador de aniones orgánicos (OATP, por sus siglas en inglés)1B1, al OATP1B3, al transportador de aniones orgánicos (OAT, por sus siglas en inglés)1, al OAT3 y al transportador de cationes orgánicos (OCT, por sus siglas en inglés)2 a concentraciones clínicamente relevantes, pero puede inhibir potencialmente a la BCRP en el tracto gastrointestinal y al OCT1.
Medicamentos antiarrítmicos y otras sustancias que pueden prolongar el intervalo QT
Bosutinib debe usarse con precaución en los pacientes que tengan o que puedan presentar una prolongación del intervalo QT, incluidos los pacientes que tomen medicamentos antiarrítmicos tales como amiodarona, disopiramida, procainamida, quinidina y sotalol, u otros medicamentos que puedan conducir a una prolongación del intervalo QT, tales como cloroquina, halofantrina, claritromicina, domperidona, haloperidol, metadona y moxifloxacino (ver sección 4.4).
4.6 - Embarazo y Lactancia de BOSULIF 400 mg Comp. recub. con película
Mujeres en edad fértil/Anticoncepción
A las mujeres en edad fértil se les debe aconsejar que utilicen un método anticonceptivo eficaz durante el tratamiento con bosutinib y durante al menos 1 mes después de la última dosis y que eviten quedarse embarazadas durante el tratamiento con bosutinib. Además, se debe advertir a estas pacientes que los vómitos o la diarrea pueden reducir la eficacia de los anticonceptivos orales ya que impiden que se absorban en su totalidad.
Embarazo
Los datos relativos al uso de bosutinib en mujeres embarazadas son limitados. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar bosutinib durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos. Si se utiliza bosutinib durante el embarazo, o si la paciente se queda embarazada durante el tratamiento con bosutinib, se la debe advertir del posible riesgo para el feto.
Lactancia
Se desconoce si bosutinib y sus metabolitos se excretan en la leche materna. En un estudio con bosutinib radiomarcado [14C] en ratas, se demostró que había excreción de radioactividad derivada de bosutinib en la leche materna (ver sección 5.3). No se puede excluir el riesgo potencial en niños lactantes. Debe interrumpirse la lactancia durante el tratamiento con bosutinib.
Fertilidad
Teniendo en cuenta los hallazgos no clínicos, bosutinib podría alterar la función reproductora y la fertilidad en los seres humanos (ver sección 5.3). A los hombres que vayan a ser tratados con bosutinib se les recomienda que soliciten información sobre la preservación del esperma antes de iniciar el tratamiento, por la posibilidad de que la fertilidad se vea disminuida por el tratamiento con bosutinib.
4.7 - Efectos sobre la capacidad de conducción de BOSULIF 400 mg Comp. recub. con película
La influencia de bosutinib sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Sin embargo, si un paciente en tratamiento con bosutinib experimenta mareo, fatiga, alteraciones visuales u otras reacciones adversas con un potencial impacto sobre la capacidad para conducir y utilizar máquinas de manera segura, el paciente debe evitar realizar estas actividades mientras persistan las reacciones adversas.
4.8 - Reacciones Adversas de BOSULIF 400 mg Comp. recub. con película
Resumen del perfil de seguridad
Un total de 1 372 pacientes con leucemia recibieron al menos una dosis de bosutinib como agente único. La mediana de la duración del tratamiento fue de 26,30 meses (intervalo: de 0,03 a 170,49 meses). Dichos pacientes tenían LMC en fase crónica recién diagnosticada, o bien LMC en fase crónica, acelerada o blástica resistente o intolerante a tratamientos anteriores, o bien tenían leucemia linfoblástica aguda (LLA) Ph+. De dichos pacientes, 268 (dosis inicial de 400 mg) y 248 (dosis inicial de 500 mg) procedían de los 2 estudios de fase 3 realizados en pacientes con LMC no tratados previamente, 60 (dosis inicial de 400 mg) procedían de un estudio de fase 2 realizado en pacientes con LMC no tratados previamente, 570 y 63 pacientes (fase 2: dosis inicial de 500 mg) procedían de 2 estudios de fase 1/2 realizados en pacientes con leucemias Ph+ tratadas previamente y 163 (dosis inicial de 500 mg) procedían de un estudio de fase 4 realizado en pacientes de LMC tratados previamente. La mediana de la duración del tratamiento fue de 55,1 meses (intervalo: de 0,2 a 60,05 meses), de 61,6 meses (intervalo: de 0,03 a 145,86 meses), de 15,3 meses (intervalo: de 0,3 a 21,8 meses), de 11,1 meses (intervalo: de 0,03 a 170,49 meses), de 30,2 meses (intervalo: de 0,2 a 85,6 meses) y de 37,80 meses (intervalo: de 0,16 a 50,0 meses), respectivamente. Los análisis de seguridad incluyeron datos de un estudio de extensión finalizado.
Se notificó al menos una reacción adversa de grado variable de toxicidad en 1 349 (98,3%) pacientes. Las reacciones adversas notificadas más frecuentemente en ≥ 20% de los pacientes fueron diarrea (80,4%), náuseas (41,5%), dolor abdominal (35,6%), trombocitopenia (34,4%), vómitos (33,7%), erupción cutánea (32,8%), ALT elevada (28,0%), anemia (27,2%), pirexia (23,4%), AST elevada (22,5%), fatiga (32,0%) y cefalea (20,3%). Se notificó al menos una reacción adversa de grado 3 o grado 4 en 943 (68,7%) pacientes. Las reacciones adversas de grado 3 o grado 4 notificadas en ≥ 5% de los pacientes fueron trombocitopenia (19,7%), ALT aumentada (14,6%), neutropenia (10,6%), diarrea (10,6%), anemia (10,3%), lipasa elevada (10,1%) y AST elevada (6,7%) y erupción cutánea (5,0%).
Tabla de reacciones adversas
Las siguientes reacciones adversas se notificaron en pacientes de los estudios clínicos con bosutinib (Tabla 2). Representan una evaluación de los datos de las reacciones adversas procedentes de 1 372 pacientes con LMC en fase crónica recién diagnosticada, con LMC en fase crónica, acelerada o blástica resistentes o intolerantes a tratamientos anteriores o con LLA Ph+ que habían recibido al menos una dosis de bosutinib como agente único.
Estas reacciones adversas se presentan conforme a la clasificación por órganos y sistemas y frecuencia. Las categorías de frecuencia se definen mediante la siguiente convención: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1 000 a < 1/100), raras (≥ 1/10 000 a < 1/1 000), muy raras (< 1/10 000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada intervalo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 2 - Reacciones adversas de bosutinib
| Infecciones e infestaciones | |
| Muy frecuentes | Infección del tracto respiratorio (incluida infección del tracto respiratorio inferior, infección viral del tracto respiratorio, infección del tracto respiratorio superior, infección viral del tracto respiratorio superior), nasofaringitis |
| Frecuentes | Neumonía (incluida neumonía atípica, neumonía bacteriana, neumonía fúngica, neumonía necrosante, neumonía estreptocócica), gripe (incluida gripe H1N1), bronquitis |
| Neoplasias benignas, malignas y no especificadas (incl quistes y pólipos) | |
| Poco frecuentes | Síndrome de lisis tumoral** |
| Trastornos de la sangre y del sistema linfático | |
| Muy frecuentes | Trombocitopenia (incluido recuento de plaquetas disminuido), neutropenia (incluido recuento de neutrófilos disminuido), anemia (incluida hemoglobina disminuida, recuento de glóbulos rojos disminuido) |
| Frecuentes | Leucopenia (incluido recuento de glóbulos blancos disminuido) |
| Poco frecuentes | Neutropenia febril, granulocitopenia |
| Trastornos del sistema inmunológico | |
| Frecuentes | Hipersensibilidad al medicamento |
| Poco frecuentes | Shock anafiláctico |
| Trastornos del metabolismo y de la nutrición | |
| Muy frecuentes | Apetito disminuido |
| Frecuentes | Deshidratación, hiperpotasemia (incluido potasio elevado en sangre), hipofosfatemia (incluido fósforo disminuido en sangre) |
| Trastornos del sistema nervioso | |
| Muy frecuentes | Mareo, cefalea |
| Frecuentes | Disgeusia |
| Trastornos del oído y del laberinto | |
| Frecuentes | Acúfenos |
| Trastornos cardiacos | |
| Frecuentes | Derrame pericárdico |
| Poco frecuentes | Pericarditis |
| Trastornos vasculares | |
| Frecuentes | Hipertensión (incluida tensión arterial aumentada, tensión arterial sistólica aumentada, hipertensión esencial, crisis hipertensiva) |
| Trastornos respiratorios, torácicos y mediastínicos | |
| Muy frecuentes | Derrame pleural, disnea, tos |
| Frecuentes | Hipertensión pulmonar (incluida hipertensión arterial pulmonar, elevación de la presión arterial pulmonar), insuficiencia respiratoria |
| Poco frecuentes | Edema pulmonar agudo (incluido edema pulmonar) |
| Frecuencia no conocida | Enfermedad pulmonar intersticial |
| Trastornos gastrointestinales | |
| Muy frecuentes | Diarrea, vómitos, náuseas, dolor abdominal (incluido malestar abdominal, dolor en la zona inferior del abdomen, dolor en la zona superior del abdomen, dolor abdominal a la palpación, dolor gastrointestinal) |
| Frecuentes | Hemorragia gastrointestinal (incluida hemorragia anal, hemorragia gástrica, hemorragia intestinal, hemorragia en el tracto gastrointestinal inferior, hemorragia rectal, hemorragia digestiva superior), pancreatitis (incluida pancreatitis aguda), gastritis |
| Trastornos hepatobiliares | |
| Frecuentes | Hepatotoxicidad (incluida hepatitis, hepatitis tóxica, trastorno hepático), función hepática anormal (incluidas enzimas hepáticas aumentadas, prueba de función hepática anormal, prueba de función hepática aumentada, transaminasas aumentadas) |
| Poco frecuentes | Lesión hepática (incluida lesión hepática inducida por el medicamento, lesión hepatocelular) |
| Trastornos de la piel y del tejido subcutáneo | |
| Muy frecuentes | Erupción (incluida erupción macular, erupción maculopapular, erupción papular, erupción pruriginosa), prurito |
| Frecuentes | Reacción de fotosensibilidad (incluida erupción polimorfa lumínica), urticaria, acné |
| Poco frecuentes | Eritema multiforme, erupción exfoliativa, erupción medicamentosa |
| Frecuencia no conocida | Síndrome de Stevens-Johnson**, necrólisis epidérmica tóxica** |
| Trastornos musculoesqueléticos y del tejido conjuntivo | |
| Muy frecuentes | Artralgia, dolor de espalda |
| Frecuentes | Mialgia |
| Trastornos renales y urinarios | |
| Frecuentes | Fallo renal agudo, fallo renal, insuficiencia renal |
| Trastornos generales y alteraciones en el lugar de administración | |
| Muy frecuentes | Edema (incluido edema palpebral, edema facial, edema generalizado, edema localizado, edema periférico, edema periorbitario, hinchazón periorbitaria, hinchazón periférica, hinchazón, hinchazón de los párpados), pirexia, fatiga (incluida astenia, malestar general) |
| Frecuentes | Dolor torácico (incluido malestar torácico), dolor |
| Exploraciones complementarias | |
| Muy frecuentes | Lipasa elevada (incluida hiperlipasemia), alanina aminotransferasa elevada (incluida alanina aminotransferasa anormal), aspartato aminotransferasa elevada, creatinina elevada en sangre |
| Frecuentes | Intervalo QT del electrocardiograma prolongado (incluido síndrome de QT largo), amilasa elevada (incluida hiperamilasemia), creatina-fosfoquinasa en sangre elevada, gamma glutamiltransferasa elevada, bilirrubina elevada en sangre (incluida hiperbilirrubinemia, bilirrubina conjugada elevada, bilirrubina no conjugada elevada en sangre) |
** Reacción adversa identificada tras la comercialización.
Descripción de reacciones adversas seleccionadas
Las descripciones que se incluyen a continuación están basadas en las observaciones realizadas sobre la seguridad de una población de 1 372 pacientes, quienes recibieron al menos una dosis de bosutinib y que o bien padecían LMC en FC recién diagnosticada, o bien LMC en FC, FA o FB resistente o intolerante a tratamientos anteriores, o bien padecían LLA Ph+.
Trastornos de la sangre y del sistema linfático
De los 372 (27,1%) pacientes con notificaciones por reacciones adversas de anemia, a 6 pacientes se les suspendió bosutinib a causa de la anemia. Se produjo toxicidad máxima de grado 1 en 95 (25,5%) pacientes, de grado 2 en 135 (36,3%) pacientes, de grado 3 en 113 pacientes (30,4%), y de grado 4 en 29 (7,8%) pacientes. En estos pacientes, la mediana del tiempo hasta el primer acontecimiento fue de 29 días (intervalo comprendido entre 1 y 3 999 días) y la mediana de la duración por acontecimiento fue de 22 días (intervalo comprendido entre 1 y 3 682 días).
De los 209 (15,2%) pacientes con notificaciones por reacciones adversas de neutropenia, a 19 pacientes se les suspendió bosutinib a causa de la neutropenia. Se produjo toxicidad máxima de grado 1 en 19 pacientes (9,1%), de grado 2 en 45 (21,5%) pacientes, de grado 3 en 95 (45,5%) pacientes, y de grado 4 en 50 (23,9%) pacientes. En estos pacientes, la mediana del tiempo hasta el primer acontecimiento fue de 56 días (intervalo comprendido entre 1 y 1 769 días) y la mediana de la duración por acontecimiento fue de 15 días (intervalo comprendido entre 1 y 913 días).
De los 472 (34,4%) pacientes con notificaciones por reacciones adversas de trombocitopenia, a 42 pacientes se les suspendió el tratamiento con bosutinib a causa de la trombocitopenia. Se produjo toxicidad máxima de grado 1 en 114 (24,2%) pacientes, de grado 2 en 88 (18,6%) pacientes, de grado 3 en 172 (36,4%) pacientes, y de grado 4 en 98 (20,8%) pacientes. En estos pacientes, la mediana del tiempo hasta el primer acontecimiento fue de 28 días (intervalo comprendido entre 1 y 1 688 días) y la mediana de la duración por acontecimiento fue de 15 días (intervalo comprendido entre 1 y 3 921 días).
Trastornos hepatobiliares
En los pacientes con notificaciones por reacciones adversas de elevaciones de ALT o de AST (todos los grados), la mediana del tiempo observado hasta la aparición de estos trastornos fue de 29 días, con un intervalo comprendido entre 1 y 3 995 días hasta la aparición de la elevación de la ALT y la AST.
La mediana de la duración por acontecimiento fue de 17 días (intervalo comprendido entre 1 y 1 148 días) y de 15 días (intervalo comprendido entre 1 y 803) para la ALT y la AST, respectivamente.
Se han producido dos casos concordantes con lesión hepática inducida por el medicamento (definida como elevaciones simultáneas de las concentraciones de ALT o AST ≥ 3 x LSN con la bilirrubina total > 2 x LSN y con la fosfatasa alcalina < 2 x LSN) sin causas alternativas en 2/1 711 (0,1%) de los sujetos tratados con bosutinib.
Reactivación del virus de la hepatitis B
Se ha notificado reactivación de la hepatitis B en relación con los TKIs BCR-ABL. En algunos casos se ha producido insuficiencia hepática aguda o hepatitis fulminante que ha dado lugar a trasplante de hígado o a un desenlace mortal (ver sección 4.4).
Trastornos gastrointestinales
De los 1 103 (80,4%) pacientes que experimentaron diarrea, a 14 se les retiró el tratamiento con bosutinib a causa de ese acontecimiento. Se administraron medicamentos concomitantes para tratar la diarrea en 756 (68,5%) pacientes. Se produjo toxicidad máxima de grado 1 en 575 (52,1%) pacientes, de grado 2 en 383 (34,7%) pacientes y de grado 3 en 144 (13,1%) pacientes; 1 paciente (0,1%) sufrió un acontecimiento de grado 4. En los pacientes con diarrea, la mediana del tiempo hasta el primer acontecimiento fue de 2 días (intervalo comprendido entre 1 y 2 702 días) y la mediana de la duración de la diarrea de cualquier grado fue de 2 días (intervalo comprendido entre 1 y 4 247 días).
De los 1 103 pacientes con diarrea, a 218 (19,8%) pacientes se les interrumpió el tratamiento, y a 208 (95,4%) de ellos se les volvió a instaurar el tratamiento con bosutinib. De los pacientes a quienes se les reinstauró el tratamiento, 201 (96,6%) no presentaron más acontecimientos diarreicos o no se les suspendió bosutinib a causa de acontecimientos diarreicos posteriores.
Trastornos cardiacos
Siete pacientes (0,5%) sufrieron una prolongación del QTcF (superior a 500 milisegundos). Once (0,8%) pacientes experimentaron un aumento del QTcF > 60 ms desde el nivel basal. En los estudios clínicos no se incluyeron pacientes con enfermedades cardiovasculares no controladas o importantes, incluyendo prolongación del QTc en los electrocardiogramas basales (ver las secciones 5.1 y 5.3).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 - Sobredosificación de BOSULIF 400 mg Comp. recub. con película
En los estudios clínicos realizados, la experiencia de sobredosis con bosutinib se limitó a casos aislados. A los pacientes que tomen una sobredosis de bosutinib se les debe mantener en observación y proporcionar el tratamiento de apoyo adecuado.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de BOSULIF 400 mg Comp. recub. con película
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores directos de la protein-quinasa, código ATC: L01EA04.
Mecanismo de acción
Bosutinib pertenece a un grupo farmacológico de medicamentos conocidos como inhibidores de las quinasas. Bosutinib inhibe la quinasa anormal BCR-ABL que promueve la LMC. Los estudios de modelización indican que bosutinib se une al dominio quinasa de BCR-ABL. Bosutinib es además un inhibidor de la familia de quinasas Src, que incluye Src, Lyn y Hck. Bosutinib inhibe mínimamente el receptor del factor de crecimiento derivado de plaquetas (PDGF, por sus siglas en inglés) y c-Kit.
En los estudios in vitro, bosutinib inhibe la proliferación y la supervivencia de líneas celulares conocidas de LMC, de líneas celulares de LLA Ph+ y de células de LMC primitivas primarias, obtenidas de pacientes. Bosutinib inhibió 16 de 18 formas de BCR-ABL resistentes a imatinib, expresadas en líneas celulares mieloides murinas. El tratamiento con bosutinib redujo el tamaño de los tumores de LMC que crecían en ratones desnudos (nude mice) e inhibió el crecimiento de tumores mieloides murinos que expresaban formas de BCR-ABL resistentes a imatinib. Además, bosutinib inhibe los receptores de tirosin-quinasa c-Fms, EphA y los receptores B, las quinasas de la familia Trk, las quinasas de la familia Axl, las quinasas de la familia Tec, algunos miembros de la familia ErbB, la tirosin-quinasa Csk no asociada a receptor, las serina/treonina quinasas de la familia Ste20 y 2 protein-quinasas dependientes de la calmodulina.
Efectos farmacodinámicos
En un estudio aleatorizado, con dosis única, doble ciego (respecto de bosutinib), cruzado, abierto, controlado con placebo y con moxifloxacino, se evaluó el efecto en sujetos sanos, de la administración de bosutinib 500 mg sobre el QTc corregido.
De los datos de este estudio se desprende que bosutinib no prolonga el QTc en sujetos sanos a una dosis de 500 mg al día con alimentos, ni en condiciones que den lugar a la elevación supraterapéutica de las concentraciones plasmáticas. Tras la administración de una dosis única, por vía oral, de bosutinib 500 mg (dosis terapéutica) y de bosutinib 500 mg junto con 400 mg de ketoconazol (para alcanzar concentraciones supraterapéuticas de bosutinib) en sujetos sanos, el límite superior del intervalo de confianza (IC) unilateral del 95% en torno al cambio promedio del intervalo QTc, fue inferior a 10 ms en todos los momentos posteriores a la administración de la dosis, y no se observaron acontecimientos adversos que pudieran sugerir una prolongación del QTc.
En un estudio realizado en sujetos con deterioro hepático, se observó una frecuencia creciente de prolongación del intervalo QTc > 450 ms, a medida que la función hepática disminuía. En el estudio clínico de fase 1/2 realizado en pacientes con leucemias Ph+ tratadas previamente tratados con bosutinib 500 mg, se observó en 9 (1,6%) de 570 pacientes, un aumento en el QTcF > 60 ms respecto del valor basal. En el estudio clínico de fase 3 realizado en pacientes con LMC en FC recién diagnosticados tratados con bosutinib 400 mg, no hubo pacientes en el grupo de tratamiento con bosutinib (N = 268) con un aumento en el QTcF > 60 ms en relación con el valor basal. En el estudio clínico de fase 3 realizado en pacientes con LMC Ph+ en FC recién diagnosticados tratados con bosutinib 500 mg, se observó en 2 (0,8%) de 248 pacientes que recibían bosutinib, un aumento en el QTcF > 60 ms respecto del valor basal. En el ensayo clínico de fase 4 en pacientes con LMC Ph+ tratados previamente con 1 o más TKI tratados con bosutinib 500 mg (N = 163), no hubo pacientes con un aumento en el QTcF > 60 ms respecto del valor basal. No se puede descartar que bosutinib tenga potencial proarrítmico.
Eficacia clínica
Estudio clínico realizado en pacientes con LMC en FC no tratados previamente
Estudio con bosutinib 400 mg
Se llevó a cabo un estudio de superioridad, multicéntrico, de dos grupos, de fase 3, abierto, para evaluar la eficacia y la seguridad de bosutinib 400 mg una vez al día comparado con imatinib 400 mg una vez al día en pacientes adultos con LMC Ph+ en FC recién diagnosticada. En el estudio se aleatorizaron 536 pacientes (268 en cada grupo de tratamiento) con LMC en FC recién diagnosticada Ph+ o Ph- (población por intención de tratar [ITT]), incluidos 487 pacientes con LMC Ph + que tenían transcritos de b2a2 y/o b3a2, y copias de BCR-ABL iniciales > 0 (población de análisis por intención de tratar modificada [ITTm, por sus siglas en inglés]).
La variable primaria de eficacia fue la proporción que mostró una respuesta molecular mayor (RMM) a los 12 meses (48 semanas) en el grupo de tratamiento con bosutinib comparado con la del grupo de tratamiento con imatinib en la población por ITTm. La RMM se definió como una proporción BCR-ABL/ABL ≤ 0,1% según la escala internacional (que corresponde a una reducción logarítmica ≥ 3 respecto al valor basal estándar) con un mínimo de 3 000 transcritos ABL según lo evaluado por el laboratorio central.
Las variables secundarias clave incluyeron la respuesta citogenética completa (RCyC) a los 12 meses, la duración de la RCyC, la duración de la RMM, la supervivencia libre de acontecimientos (SLA) y la supervivencia global (SG). La RCyC en el mes 12 se definió como la ausencia de metafases en el Ph+ en el bandeo cromosómico de ≥ 20 metafases obtenidas del aspirado de médula ósea o una RMM si no se disponía de una evaluación citogenética adecuada. Los valores-p para variables distintas de la RMM a los 12 meses y la RCyC a los 12 meses no se han ajustado para comparaciones múltiples.
Las características basales para la población por ITTm estaban bien equilibradas entre los 2 grupos de tratamiento con respecto a la edad (la mediana de edad fue de 52 años para el grupo de bosutinib y de 53 años para el grupo de imatinib con el 19,5% y el 17,4% de los pacientes con 65 años o más, respectivamente); sexo (42,3% y 44,0% de mujeres, respectivamente); raza (78,0% y 77,6% de raza caucásica, 12,2% y 12,4% de raza asiática, 4,1% y 4,1 % de raza negra o afroamericana, y 5,7% y 5,4% de otras razas, respectivamente, además de 1 con raza desconocida en el grupo de imatinib); y puntuación de riesgo de Sokal (riesgo bajo 35,0% y 39,4%, riesgo intermedio 43,5% y 38,2%, riesgo alto 21,5% y 22,4%, respectivamente).
Después de 60 meses de seguimiento en la población por ITTm, el 60,2% de los pacientes tratados con bosutinib (N = 246) y el 59,8% de los pacientes tratados con imatinib (N = 239) seguían recibiendo tratamiento de primera línea.
Después de 60 meses de seguimiento en la población por ITTm, las interrupciones debidas a la progresión de la enfermedad a LMC en FA o en FB para los pacientes tratados con bosutinib fueron del 0,8% comparado con el 1,7% de los pacientes tratados con imatinib. En 6 (2,4%) pacientes con bosutinib y 7 (2,9%) pacientes con imatinib tuvieron lugar transformaciones a LMC en FA o LMC en FB. Las interrupciones debidas a una respuesta insuficiente o al fracaso del tratamiento según lo evaluado por el investigador ocurrieron en el 5,3% de los pacientes en el grupo tratado con bosutinib comparado con el 15,5% de los pacientes en el grupo tratado con imatinib. Doce (4,9%) pacientes con bosutinib y 14 (5,8) pacientes con imatinib murieron mientras estaban en el estudio. No se produjeron transformaciones adicionales en la población por ITT, hubo 2 muertes adicionales en el grupo de bosutinib en la población por ITT.
Los resultados de eficacia de la RMM y la RCyC se resumen en la Tabla 3.
Tabla 3 - Resumen de la RMM a los 12 y 18 meses y la RCyC a los 12 meses, por grupo de tratamiento en la población por ITTm
Respuesta | Bosutinib (N=246) | Imatinib (N=241) | Cociente de probabilidad (Odds ratio) (IC del 95%)a |
| (IC del 95%) | (40,9; 53,4) | (30,8; 43,0) | |
| Valor-p unilateral | 0,0100b | ||
| (IC del 95%) | (50,7; 63,1) | (41,4; 54,0) | |
| Valor-p unilateral | 0,0208c | ||
| (IC del 95%) | (72,0; 82,5) | (60,4; 72,4) | |
| Valor-p unilateral | 0,0037b | ||
Nota: La respuesta molecular mayor se definió como una proporción BCR-ABL/ABL ≤ 0,1% según la escala internacional (que corresponde a una reducción logarítmica ≥ 3 respecto al valor basal estándar) con un mínimo de 3 000 transcritos ABL según lo evaluado por el laboratorio central. La respuesta citogenética completa se definió como la ausencia de metafases en el Ph+ en el bandeo cromosómico de ≥ 20 metafases obtenidas del aspirado de médula ósea o una RMM si no se disponía de una evaluación citogenética adecuada.
Abreviaturas: BCR-ABL = breakpoint cluster region-Abelson; IC = intervalo de confianza; CMH = Cochran-Mantel-Haenszel; RCyC = respuesta citogenética completa; ITTm = intención de tratar modificada; RMM = respuesta molecular mayor; N/n = número de pacientes; Ph+ = cromosoma Philadelphia positivo.
a Ajustado por región geográfica y puntuación de Sokal en la aleatorización.
b Comparación estadísticamente significativa en el nivel de significancia especificado previamente; basada en la prueba de CMH estratificada por región geográfica y la puntuación de Sokal en la aleatorización.
c Basada en la prueba de CMH estratificada por región geográfica y la puntuación de Sokal en la aleatorización.
En el mes 12, la tasa de RM4 (definida como ≤ 0,1% BCR-ABL [que corresponde a una reducción logarítmica ≥ 4 respecto al valor basal estándar] con un mínimo de 9 800 transcritos) fue mayor en el grupo de tratamiento con bosutinib comparado con el grupo de tratamiento con imatinib en la población por ITTm (20,7% [IC del 95%: 15,7%; 25,8%] frente al 12,0% [IC del 95%: 7,9%; 16,1%], respectivamente, cociente de probabilidad (Odds ratio, OR, por sus siglas en inglés) 1,88 [IC del 95%: 1,15; 3,08], valor-p unilateral = 0,0052).
En los meses 3, 6 y 9, la proporción de pacientes con RMM fue más alta en el grupo de tratamiento con bosutinib comparado con el grupo de tratamiento con imatinib (Tabla 4).
Tabla 4 - Comparación de la RMM en los meses 3, 6 y 9 por tratamiento en la población por ITTm
| Tiempo | Número (%) de sujetos con RMM | Cociente de probabilidad (Odds ratio) (IC del 95%)a | |
| Bosutinib (N=246) | Imatinib (N=241) | ||
| (IC del 95%) | (1,6; 6,5) | (0,0; 3,3) | |
| Valor-p unilateralb | 0,0578 | ||
| (IC del 95%) | (29,0; 40,9) | (13,4; 23,1) | |
| Valor-p unilateralb | < 0,0001 | ||
| (IC del 95%) | (36,1; 48,4) | (23,7; 35,2) | |
| Valor-p unilateralb | 0,0015 | ||
Nota: Los porcentajes se basaron en el número de pacientes en cada grupo de tratamiento. RMM se definió como una proporción BCR-ABL/ABL ≤ 0,1% según la escala internacional (que corresponde a una reducción logarítmica ≥ 3 respecto al valor basal estándar) con un mínimo de 3 000 transcritos ABL según lo evaluado por el laboratorio central.
Abreviaturas: BCR-ABL = breakpoint cluster region-Abelson; IC = intervalo de confianza; CMH = Cochran-Mantel-Haenszel; ITTm = intención de tratar modificada; RMM = respuesta molecular mayor; N = número de pacientes.
a Ajustado por región geográfica y puntuación de Sokal en la aleatorización.
b Basado en la prueba de CMH estratificada por región geográfica y la puntuación de Sokal en la aleatorización.
A los 60 meses, en la población por ITTm, la proporción de pacientes con RMM, RM4 y RM4,5 fue mayor en el grupo de bosutinib en comparación con el grupo de imatinib (Tabla 5). Las tasas de RMM a los 60 meses en los subgrupos de riesgo de Sokal se resumen en la Tabla 6.
Tabla 5 - Resumen de la respuesta molecular a los 60 meses en la población por ITTm
| Respuesta | Bosutinib (N = 246) | Imatinib (N = 241) | Cociente de probabilidad (Odds ratio) (IC del 95%)a |
| Respuesta molecular a los 60 meses, n (%) (IC del 95%) | |||
| RMM | 182 (74,0) (68,5; 79,5) | 158 (65,6) (59,6; 71,6) | 1,52 (1,02; 2,25) |
| RM4 | 145 (58,9) (52,8; 65,1) | 120 (49,8) (43,5; 56,1) | 1,46 (1,02; 2,09) |
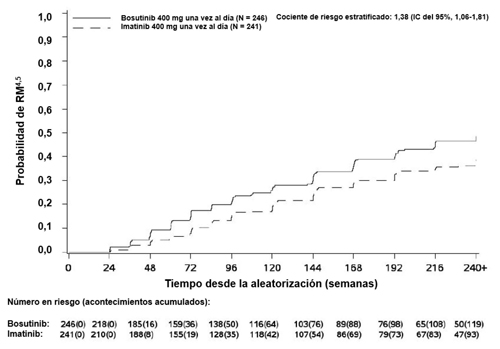
| RM4,5 | 119 (48,4) (42,1; 54,6) | 93 (38,6) (32,4; 44,7) | 1,50 (1,05; 2,16) |
Nota: RMM/RM4/RM4,5 se definieron como una proporción BCR-ABL/ABL ≤ 0,1/0,01/0,0032% según la escala internacional (que corresponde a una reducción logarítmica ≥ 3/4/4,5 respecto al valor basal estándar) con un mínimo de 3 000/9 800/30 990 transcritos ABL según lo evaluado por el laboratorio central.
Abreviaturas: BCR-ABL = breakpoint cluster region-Abelson; IC = intervalo de confianza; ITTm = intención de tratar modificada; RMM = respuesta molecular mayor; RM = respuesta molecular; N/n = número de pacientes.
a Ajustado por región geográfica y puntuación de Sokal en la aleatorización.
Tabla 6 - Resumen de la RMM a los 60 meses por la puntuación de riesgo de Sokal en la población por ITTm
| Respuesta | Bosutinib | Imatinib | Cociente de probabilidad (Odds ratio) (IC del 95%) |
| (IC del 95%) | (69,1; 86,7) | (62,5; 80,6) | |
| (IC del 95%) | (65,5; 82,2) | (57,8; 77,0) | |
| (IC del 95%) | (55,4; 80,5) | (38,5; 65,2) |
Nota: Los porcentajes se basaron en el número de pacientes en cada grupo de tratamiento. RMM se definió como una proporción BCR-ABL/ABL ≤ 0,1% según la escala internacional (que corresponde a una reducción logarítmica ≥ 3 respecto al valor basal estándar) con un mínimo de 3 000 transcritos ABL según lo evaluado por el laboratorio central.
Abreviaturas: BCR-ABL = breakpoint cluster region-Abelson; IC = intervalo de confianza; ITTm = intención de tratar modificada; RMM = respuesta molecular mayor; N/n = número de pacientes.
La incidencia acumulada de la RCyC ajustada por riesgo competitivo de interrupción del tratamiento sin RCyC fue mayor en el grupo de tratamiento con bosutinib comparado con el grupo de tratamiento con imatinib en la población por ITTm (83,3% [IC del 95%: 78,1%; 87,4%] frente al 76,8% [IC del 95%: 70,9%; 81,6%] en el mes 60; cociente de riesgo [CR] o Hazard Ratio [HR, por sus siglas en inglés] de un modelo estratificado de riesgos subdistribucionales proporcionales: 1,35; [IC del 95%: 1,11; 1,64]). La mediana del tiempo hasta la RCyC (solo en los respondedores) fue de 24,0 semanas (intervalo: de 11,4 a 120,7) en el grupo de bosutinib comparado con 24,3 semanas en el grupo de imatinib (intervalo: de 11,4 a 96,6).
La mediana de tiempo hasta la RMM, RM4 y RM4,5 (solo en los respondedores) fue de 36,1 semanas (intervalo: de 11,9 a 241,9), 83,7 semanas (intervalo: de 12,4 a 244,3) y 108,0 semanas (intervalo: de 24,1 a 242,1), respectivamente, para el grupo de tratamiento con bosutinib frente a 47,7 semanas (intervalo: de 12,1 a 216,1), 84,4 semanas (intervalo: de 23,6 a 241,9) y 120,4 semanas (intervalo: de 24,6 a 240,7), respectivamente, para el grupo de tratamiento con imatinib en la población por ITTm.
La incidencia acumulada de RMM, RM4 y RM4,5 ajustada por riesgo competitivo de la interrupción del tratamiento sin acontecimientos fue mayor con bosutinib en comparación con imatinib, como se muestra en las Figuras 1 a 3.
Figura 1 – Incidencia acumulada de RMM (población por ITTm)
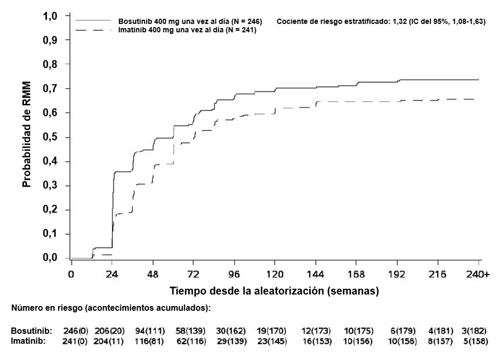
Figura 2 – Incidencia acumulada de RM4 (población por ITTm)
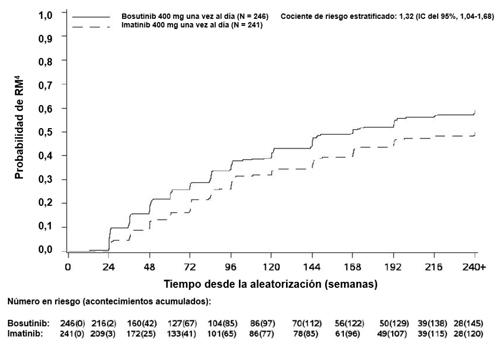
Figura 3 – Incidencia acumulada de RM4,5 (población por ITTm)
En la población por ITTm, entre los pacientes que alcanzaron RCyC, la estimación de mantener una respuesta en el año 4 usando el método de Kaplan-Meier fue del 97,4% (IC del 95%: 93,9%; 98,9%) y 93,7% (IC del 95%: 88,9%; 96,5%) en los grupos de bosutinib e imatinib (CR 0,39 [IC del 95%: 0,14; 1,13]), respectivamente. Entre los pacientes que alcanzaron la RMM, la estimación de mantener una respuesta en el año 4 usando el método de Kaplan-Meier fue del 92,2% (IC del 95%: 86,8%; 95,4%) y del 92,0% (IC del 95%: 85,9%; 95,5%) en los grupos de bosutinib e imatinib (CR 1,09 [IC del 95%: 0,49; 2,44]), respectivamente.
A los 60 meses, el 43,9% (IC del 95%: 37,7%; 50,1%) y el 38,6% (IC del 95%: 32,4%; 44,7%) de los pacientes tratados con bosutinib e imatinib (OR 1,24 [IC del 95%: 0,87; 1,78]) en la población por ITTm, respectivamente, presentaban una RM4 continuada definida por los siguientes criterios: tratamiento durante al menos 3 años con al menos RM4 en todas las evaluaciones durante un período de 1 año.
La incidencia acumulada de SLA durante el tratamiento en el mes 60 en la población por ITTm fue del 6,9% (IC del 95%: 4,2%; 10,5%) en el grupo de bosutinib y del 10,4% (IC del 95%: 6,9%; 14,6%) en el grupo de imatinib (CR 0,64; IC del 95%: 0,35; 1,17).
Las estimaciones de la SG usando el método de Kaplan-Meier en el mes 60 para los pacientes tratados con bosutinib e imatinib en la población por ITTm fueron del 94,9% (IC del 95%: 91,1%; 97,0%) y 94,0% (IC del 95%: 90,1%; 96,4%), respectivamente (CR 0,80; IC del 95%: 0,37; 1,73).
En un análisis retrospectivo, entre los pacientes evaluables en la población por ITT, más pacientes en el grupo de bosutinib 200/248 (80,6%) alcanzaron una respuesta molecular temprana (transcritos de BCR-ABL ≤ 10% a los 3 meses) comparado con los pacientes del grupo de imatinib 153/253 (60,5%), OR 2,72 (IC del 95%: 1,82; 4,08). La RMM y la SLA en el mes 60 en pacientes tratados con bosutinib con y sin respuesta molecular temprana se resumen en la Tabla 7.
Tabla 7 - Resultados en el mes 60 en pacientes tratados con bosutinib con BCR-ABL ≤ 10% frente a > 10% en el mes 3 en la población por ITT
| Bosutinib (N = 248) | Pacientes con BCR-ABL ≤ 10% a los 3 meses (N = 200) | Pacientes con BCR-ABL > 10% a los 3 meses (N = 48) | Cociente de riesgo (Hazard Ratio) (IC del 95%)a |
| Incidencia acumulada de RMM, % (IC del 95%) | 84,0 (78,1; 88,4) | 56,5 (41,1; 69,4) | 2,67 (1,90; 3,75) |
| Incidencia acumulada de SLA, % (IC del 95%) | 5,5 (2,9; 9,3) | 12,5 (5,1; 23,4) | 0,40 (0,14; 1,17) |
Abreviaturas: BCR-ABL = breakpoint cluster region-Abelson; IC = intervalo de confianza; ITT = intención de tratar; RMM = respuesta molecular mayor; SLA: supervivencia libre de acontecimientos; N = número de pacientes con ≥ 3 000 copias de ABL en el mes 30.
a Ajustado por región geográfica y puntuación de Sokal en la aleatorización.
Menos pacientes en el grupo de bosutinib [6 (2,4%) bosutinib y 12 (5,0%) imatinib] presentaron nuevas mutaciones detectables a los 60 meses en la población por ITTm.
Estudio clínico de fase 1/2 realizado en pacientes con LMC en FC, FA y FB resistente o con intolerancia a imatinib
Se llevó a cabo un estudio de fase 1/2, multicéntrico, abierto, de un único grupo para evaluar la eficacia y la seguridad de bosutinib 500 mg una vez al día, en pacientes con LMC resistente a imatinib o con intolerancia al mismo, tratados previamente con un TKI (imatinib) o con más de un TKI (imatinib seguido de dasatinib y/o nilotinib), empleando cohortes independientes para la enfermedad en fase crónica, acelerada y blástica.
En este estudio hubo 570 pacientes tratados con bosutinib, incluyendo pacientes con LMC en fase crónica tratados previamente con un único TKI (imatinib), pacientes con LMC en fase crónica tratados previamente con imatinib y con al menos un TKI adicional (dasatinib y/o nilotinib), pacientes con LMC en fase acelerada o blástica tratados previamente con al menos un TKI (imatinib) y pacientes con LLA Ph+ tratados previamente con al menos un TKI (imatinib).
La variable primaria de eficacia del estudio fue la tasa de respuesta citogenética mayor (RCyM) en la semana 24 en los pacientes con LMC en fase crónica resistente a imatinib, tratados previamente con un único TKI (imatinib). Otras variables de eficacia incluyeron las tasas de respuesta citogenética y molecular acumuladas, el tiempo y la duración de las respuestas citogenéticas y moleculares, la respuesta en mutaciones basales, la transformación a FA/FB , la supervivencia libre de progresión y la SG en todas las cohortes.
Los pacientes que todavía estaban recibiendo bosutinib al final del estudio de fase 1/2 y se estaban beneficiando del tratamiento con bosutinib a juicio del investigador, así como aquellos pacientes que ya habían suspendido el tratamiento con bosutinib como parte del estudio de fase 1/2 y estaban en seguimiento a largo plazo para la supervivencia o habían finalizado el estudio de fase 1/2, fueron elegibles para participar en el estudio de extensión. Cada paciente permaneció en el estudio de extensión, o bien en tratamiento con bosutinib o en el seguimiento de la supervivencia a largo plazo, hasta que el último paciente alcanzó los 10 años de seguimiento, calculados a partir de la fecha de su primera dosis de bosutinib recibida en el estudio de fase 1/2.
Las variables de eficacia del estudio de extensión incluyeron la duración de las respuestas citogenéticas y moleculares, la transformación a FA/FB, la supervivencia libre de progresión y la SG.
Los análisis de eficacia incluyeron datos de este estudio de extensión finalizado.
Pacientes con LMC en FC
En la Tabla 8 se presentan los resultados de eficacia correspondientes a los pacientes con LMC Ph+ en fase crónica, tratados previamente con imatinib y al menos con un TKI adicional (seguimiento mínimo de 120 meses, una mediana de duración de tratamiento de 9 meses [intervalo: de 0,23 a 164,28 meses] y un 20,2% y 7,6% todavía en tratamiento a los 60 y 120 meses, respectivamente) y los resultados de pacientes con LMC Ph+ en fase crónica tratados previamente con imatinib únicamente (seguimiento mínimo de 120 meses, una mediana de duración de tratamiento de 26 meses [intervalo: de 0,16 a 170,49 meses] y un 40,5% y 19,4% todavía en tratamiento a los 60 y 120 meses, respectivamente).
Pacientes con LMC en FA y FB
Los resultados de eficacia correspondientes a los pacientes con LMC Ph+ en FA (seguimiento mínimo de 120 meses, una mediana de duración de tratamiento de 10 meses [intervalo: de 0,10 a 156,15 meses] y un 12,7 y 7,6% todavía en tratamiento a los 60 y 120 meses, respectivamente) y en FB (seguimiento mínimo de 120 meses, una mediana de duración de tratamiento de 2,8 meses [intervalo: de 0,03 a 71,38 meses] y un 3,1% y 0% todavía en tratamiento a los 60 y 120 meses, respectivamente) se presentan en la Tabla 8.
Tabla 8 – Resultados de eficacia en pacientes con LMC en fase crónica y en fase avanzada previamente tratados*
| LMC Ph+ en fase crónica (FC) con tratamiento previo solo con imatinib | LMC Ph+ en FC con tratamiento previo con imatinib y dasatinib o nilotinib | Fase acelerada (FA) con tratamiento previo, como mínimo, con imatinib | Fase blástica (FB) con tratamiento previo, como mínimo, con imatinib | |
| RCyC, % (IC del 95%) | 49,6 (43,4; 55,8) | 32,1 (23,6; 41,6) | 30,6 (20,2; 42,5) | 27,8 (16,5; 41,6) |
| RM4, % (IC del 95%) | 37,1 (30,3; 44,2) | 15,0 (8,8; 23,1) | 13,0 (5,4; 24,9) | 10,4 (3,5; 22,7) |
| Tiempo hasta la RCyM solo para respondedoresb, mediana (intervalo), semanas | 12,3 (4,0; 346,0) | 12,3 (3,9; 550,6) | 12,0 (3,9; 144,7) | 8,2 (3,9; 25,1) |
| Mediana, semanas (IC del 95%) | N/R | N/R | 84,0 (24,0; N/E) | 29,1 (11,9; 38,3) |
| Tiempo hasta la RCyC solo para respondedoresb, mediana (intervalo), semanas | 24,0 (7,7; 240,6) | 24,0 (11,6; 216,0) | 23,8 (4,1; 120,0) | 8,4 (3,9; 25,1) |
| Mediana, semanas (IC del 95%) | N/R | 252,0 (24,0; N/E) | 72,0 (36,1; N/E) | 20,0 (9,1; 29,6) |
| Tiempo hasta la RMM solo para respondedoresb, mediana (intervalo), semanas | 35,6 (3,1; 367,1) | 12,4 (4,0; 171,7) | 36,1 (12,1; 144,1) | 4,7 (3,9;168,9) |
| Mediana, semanas (IC del 95%) | N/R | N/R | N/R | N/R |
| Tiempo hasta la RM4 solo para respondedoresb, mediana (intervalo), semanas | 28,0 (3,1; 583,1) | 23,8 (4,0; 240,1) | 24,1 (22,9; 96,0) | 4,7 (3,9; 284,9) |
| Mediana, semanas (IC del 95%) | N/R | |||
| Transformación durante el tratamiento, n | 15 | 5 | 3 | |
| IncAcu en el año 10, % (IC del 95%)d | 23,9 (19,5; 29,5) | 26,9 (20,0; 36,2) | 41,8 (32,2; 54,2) | N/E |
| Mediana, meses (IC del 95%) | N/R | N/R | N/R | 10,9 (8,7; 19,7) |
Fecha de la recopilación de datos: estudio de fase 1/2, 02 de octubre de 2015; estudio de extensión, 02 de septiembre de 2020.
Criterios aplicados a la respuesta citogenética: la RCyM incluyó las respuestas citogenéticas completas [0% de metafases Ph+ de médula ósea o < 1% de células positivas de hibridación in situ fluorescente (FISH)] o parciales (1%-35%). Las respuestas citogenéticas se basaron en el porcentaje de metafases Ph+ de ≥ 20 células en metafase en cada muestra de médula ósea. Si no se disponía de ≥ 20 metafases para realizar las evaluaciones citogenéticas posteriores a la evaluación basal se podía usar el análisis FISH (≥ 200 células). En el estudio de extensión, la RCyC se imputó a partir de la RMM si no se disponía de una evaluación citogenética válida en una fecha específica.
Criterios aplicados a la respuesta molecular: en el estudio de fase 1/2, RMM/RM4 se definió como ≤0,1/0,01% de transcritos de BCR-ABL según lo evaluado por el laboratorio central (no según la escala internacional). En el estudio de extensión, a los respondedores se les indicó RMM/RM4 en el cuaderno de recogida de datos según lo evaluado por un laboratorio local.
Abreviaturas: FA = fase acelerada; FB = fase blástica; Ph+ = cromosoma Philadelphia positivo; FC = fase crónica; LMC = leucemia mieloide crónica; K-M = Kaplan-Meier, N/n = número de pacientes; N/A = No aplicable, N/R = No alcanzado en el tiempo mínimo de seguimiento, N/E = No estimable, IC = intervalo de confianza, RCyM = respuesta citogenética mayor, RCyC = respuesta citogenética completa, IncAcu = incidencia acumulada, RMM = respuesta molecular mayor, BCR-ABL = breakpoint cluster region Abelson.
a Incluye a los pacientes (N) con una evaluación basal válida para la respuesta citogenética y pacientes que no eran de China, Sudáfrica, India o Rusia para la respuesta molecular, ya que las muestras no se pudieron exportar para la evaluación molecular en esos países. Los resultados de los análisis permiten incluir como pacientes respondedores a aquellos que respondieron en la evaluación basal y que mantuvieron la respuesta tras ese momento. Tiempo mínimo de seguimiento (tiempo desde que se administró al último paciente su primera dosis hasta la fecha en que se recopilaron los datos) de 120 meses.
b Incluye a los pacientes (N) que lograron y mantuvieron una respuesta.
c Incluye a los pacientes (N) que recibieron al menos 1 dosis de bosutinib.
d Análisis de incidencia acumulada que se ajusta al riesgo competitivo de la interrupción del tratamiento sin el acontecimiento.
e No analizado para grupos con números limitados.
La supervivencia global en las cohortes FC, FA y FB se muestra gráficamente en la Figura 4 .
Figura 4 - Estimación de Kaplan-Meier de la supervivencia global (SG) en FC2L, FC3L, FA y FB
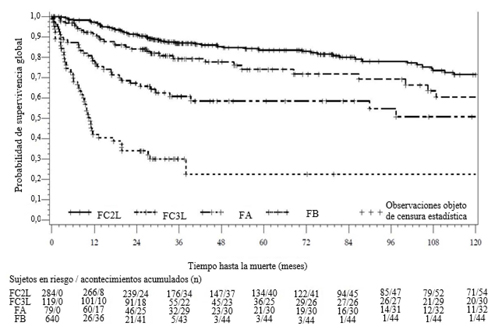
Tabla 9 – Respuesta en función del estado basal de mutación BCR-ABL en población evaluable con LMC en FC: tratamiento previo con imatinib y dasatinib y/o nilotinib (tercera línea)
| Estado basal de mutación BCR-ABL | Incidencia en el momento basal n (%)a | RCyM alcanzada o mantenida Resp/Evalb (%) N=112 |
| Mutación evaluada | 98 (100,0) | 36/92 (39,1) |
| No mutación | 59 (60,2) | 23/55 (41,8) |
| Al menos 1 mutación | 39 (39,8) | 13/37 (35,1) |
| Mutaciones resistentes a dasatinib | 10 (10,2) | 1/9 (11,1) |
| E255K/V | 2 (2,0) | 0/2 |
| F317L | 8 (8,2) | 1/7 (14,3) |
| Mutaciones resistentes a nilotinibc | 13 (13,3) | 8/13 (61,5) |
| Y253H | 6 (6,1) | 5/6 (83,3) |
| E255K/V | 2 (2,0) | 0/2 |
| F359C/I/V | 7 (7,1) | 5/7 (71,4) |
Fecha de la recopilación de datos: estudio de fase 1/2, 02 de octubre de 2015; estudio de extensión, 02 de septiembre de 2020
Nota: Las mutaciones basales se identificaron antes de la administración al paciente de la primera dosis del medicamento en investigación.
Abreviaturas: BCR-ABL = breakpoint cluster region-Abelson; FC = fase crónica; LMC = leucemia mieloide crónica; RCyM = respuesta citogenética mayor; N/n = número de pacientes; Resp=respondedores, Eval = evaluable.
a El porcentaje se basa en el número de pacientes con evaluación de mutación basal.
b La población evaluable incluye pacientes que tuvieron una evaluación válida de la enfermedad basal.
c Dos pacientes tuvieron más de 1 mutación en esta categoría.
Un paciente con la mutación E255V, tratado previamente con nilotinib, alcanzó una RHC como mejor respuesta.
Los ensayos in vitro mostraron que bosutinib tuvo una actividad limitada frente a las mutaciones T315I o V299L. Por lo tanto, no es de esperar que haya actividad clínica en pacientes con estas mutaciones.
Estudio clínico de fase 4 realizado en pacientes con LMC Ph+ previamente tratada con 1 o más TKI
Se llevó a cabo un estudio de fase 4, multicéntrico, abierto, no aleatorizado y de un único grupo para evaluar la eficacia y la seguridad de bosutinib 500 mg una vez al día en pacientes con LMC resistentes a TKI o con intolerancia a TKI empleando cohortes independientes para la enfermedad en FC, FA o FB previamente tratada con uno o más TKI.
En este estudio hubo 163 pacientes tratados con bosutinib, incluidos 46 pacientes con LMC Ph+ en FC y tratados previamente con 1 TKI (imatinib o dasatinib o nilotinib), 61 pacientes con LMC Ph+ en FC tratados previamente con 2 TKI (imatinib y/o dasatinib y/o nilotinib), 49 pacientes con LMC Ph+ en FC tratados previamente con 3 TKI (imatinib y dasatinib y nilotinib), 4 pacientes con LMC Ph+ en FA tratados previamente con al menos 1 TKI (2 pacientes tratados con 2 TKI previos y 2 pacientes tratados con 3 TKI previos) y 3 pacientes con LMC Ph− tratados con al menos 1 TKI previo.
La variable primaria de eficacia del estudio fue la RCyM acumulada confirmada al año (semana 52) en pacientes con LMC Ph+ en FC tratados previamente con 1 o 2 TKI y pacientes con LMC Ph+ en FC tratados previamente con 3 TKI. Para los pacientes con LMC Ph+ en FA y FB con cualquier tratamiento previo con TKI, la variable primaria de eficacia fue la respuesta hematológica global (RHG) acumulada confirmada al año (semana 52). Otras variables de eficacia en pacientes con LMC Ph+ en FC incluyeron la respuesta citogenética y molecular acumulada, la duración de las respuestas citogenéticas y moleculares, la respuesta en mutaciones basales, la transformación a FA/FB, la supervivencia libre de progresión (SLP) y la SG. Las variables adicionales en la cohorte Ph+ FA/FB incluyeron las tasas acumuladas de respuestas citogenéticas y moleculares, la SLP y la SG.
Pacientes con LMC en FC
La variable primaria de la tasa acumulada de RCyM confirmada (IC del 95%) al año (52 semanas) fue del 76,5% (66,9; 84,5) en los pacientes tratados con 1 o 2 TKI previos y del 62,2% (46,5; 76,2) en los pacientes tratados con 3 TKI previos.
Los resultados de eficacia adicionales al cierre del estudio, después de un seguimiento mínimo de 3 años, en pacientes con LMC Ph+ en FC tratados con 1 (mediana de la duración del tratamiento de 47,5 meses [intervalo: de 0,9 a 50,1 meses] y 60,9% todavía en tratamiento), 2 (mediana de la duración del tratamiento de 41,9 meses [intervalo: de 0,4 a 48,9 meses] y 45,9% todavía en tratamiento) y 3 (mediana de la duración del tratamiento de 20,0 meses [intervalo: de 0,2 a 48,9 meses] y 38,8% todavía en tratamiento) TKI previos se presentan en la Tabla 10.
Tabla 10 – Resultados de eficacia en pacientes con LMC Ph+ en fase crónica previamente tratados
| LMC Ph+ en FC tratada con 1 TKI previo | LMC Ph+ en FC tratada con 2 TKI previos | LMC Ph+ en FC tratada con 3 TKI previos | Cohorte total de LMC Ph+ en FC | |
| RCyMa acumulada confirmada al año, % (IC del 95%) | N=43 83,7 (69,3; 93,2) | N=55 70,9 (57,1; 82,4) | N=45 62,2 (46,5; 76,2) | N=143 72,0 (63,9; 79,2) |
RCyC, % (IC del 95%) | 86,0 (72,1; 94,7) | 83,6 (71,2; 92,2) | 73,3 (58,1; 85,4) | 81,1 (73,7; 87,2) |
RM4.5, % (IC del 95%) | 58,7 (43,2; 73,0) | 50,9 (37,1; 64,6) | 35,4 (22,2; 50,5) | 48,3 (40,1; 56,6) |
RCyC | 3,0 (1,0; 17,6) | 2,9 (0,3; 6,4) | 3,0 (1,8; 8,8) | 3,0 (0,3; 17,6) |
RCyC, K-M en el año 3, % (IC del 95%) | 96,4 (77,2; 99,5) | 94,4 (79,2; 98,6) | 100,0 (100,0; 100,0) | 96,5 (89,5; 98,9) |
RM4.5 | 9,2 (2,8; 47,6) | 6,0 (2,8; 36,2) | 5,8 (1,8; 18,0) | 6,0 (1,8; 47,6) |
RM4, K-M en el año 3, % (IC del 95%) | 89,5 (70,9; 96,5) | 68,7 (48,0; 82,5) | 85,2 (51,9; 96,2) | 80,7 (69,4; 88,1) |
| Fecha de la recopilación de datos: 23 de noviembre de 2020. Abreviaturas: Ph+ = cromosoma Philadelphia positivo; FC = fase crónica; LMC = leucemia mieloide crónica; K-M = Kaplan-Meier; N = número de pacientes; IC = intervalo de confianza; RCyM = respuesta citogenética mayor, RCyC = respuesta citogenética completa; RMM = respuesta molecular mayor; RM4 = reducción logarítmica ≥ 4 de transcritos de BCR-ABL respecto al valor basal estándar; RM4,5 = reducción logarítmica ≥ 4,5 de transcritos de BCR-ABL respecto al valor basal estándar. Criterios aplicados a la RCyM acumulada confirmada: La respuesta se confirmó con 2 evaluaciones consecutivas con al menos 28 días de diferencia. Para ser considerado un respondedor, el paciente debe haber mantenido una respuesta inicial durante al menos 52 semanas o haber mejorado con respecto al valor basal. Los pacientes con una respuesta citogenética parcial (RCyP) al inicio del tratamiento deben lograr una RCyC en el tratamiento para ser considerados respondedores citogenéticos. Los pacientes con al menos una RMM y una respuesta molecular más profunda que la basal se consideran con una RCyC confirmada. Criterios aplicados a la respuesta citogenética acumulada: La respuesta citogenética mayor incluyó respuestas citogenéticas completas [0% de metafases Ph+ de médula ósea o < 1% de células positivas de hibridación in situ fluorescente (FISH)] o parciales (1%-35%). Las respuestas citogenéticas se basaron en el porcentaje de metafases Ph+ de ≥ 20 células en metafase en cada muestra de médula ósea. Si no se disponía de ≥ 20 metafases para evaluar la RCyC se podía usar el análisis FISH (≥ 200 células). Los pacientes sin una evaluación válida de médula ósea o FISH y con al menos una RMM se consideraron con RCyC. Criterios aplicados a la respuesta molecular acumulada: RMM, RM4 y RM4,5 se definieron como ≤ 0,1%, ≤ 0,01% y ≤ 0,0032% del cociente BCR-ABL/ABL en la escala internacional, respectivamente (correspondientes a una reducción logarítmica ≥ 3, ≥ 4 y ≥ 4,5 del valor basal estándar) con un mínimo de 10.000, 10.000 y 32.000 transcritos de ABL evaluados por el laboratorio central, respectivamente. a Incluye a los pacientes (N) con una evaluación basal válida. Tiempo mínimo de seguimiento (tiempo desde que se administró al último paciente su primera dosis hasta la fecha en que se recopilaron los datos) de 36 meses. b Incluye a los pacientes (N) que lograron o mantuvieron la respuesta. | ||||
La incidencia acumulada de RMM, RM4 y RM4,5 ajustada al riesgo competitivo de la interrupción del tratamiento sin el acontecimiento se muestra en la Figura 5.
Figura 5 – Incidencia acumulada de la respuesta molecular (población en FC evaluable)
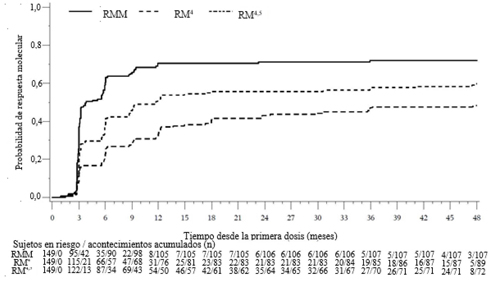
Las respuestas moleculares logradas por línea de tratamiento se muestran en la Tabla 11.
Tabla 11 – Respuestas moleculares logradas
| LMC Ph+ en FC tratada con 1 TKI previo | LMC Ph+ en FC tratada con 2 TKI previos | LMC Ph+ en FC tratada con 3 TKI previos | Cohorte total de LMC Ph+ en FC | |
RMM, % (IC del 95%) | 76,0 (54,9; 90,6) | 64,3 (44,1; 81,4) | 38,5 (20,2; 59,4) | 59,5 (47,9; 70,4) |
RM4, % (IC del 95%) | 70,3 (53,0, 84,1) | 55,3 (38,3; 71,4) | 32,4 (18,0; 49,8) | 52,7 (43,0; 62,2) |
RM4,5, % (IC del 95%) | 54,8 (38,7; 70,2) | 43,5 (28,9; 58,9) | 30,2 (17,2; 46,1) | 42,7 (34,1; 51,7) |
RM profunda, % (IC del 95%) | 85,7 (63,7; 97,0) | 66,7 (46,0; 83,5) | 63,6 (40,7; 82,8) | 71,4 (59,4; 81,6) |
| Fecha de la recopilación de datos: 23 de noviembre de 2020. Abreviaturas: Ph+ = cromosoma Philadelphia positivo; FC = fase crónica; LMC = leucemia mieloide crónica; K- M = Kaplan-Meier; N = número de pacientes; IC = intervalo de confianza; RMM = respuesta molecular mayor; RM = respuesta molecular; RM4 = reducción logarítmica ≥ 4 de transcritos de BCR-ABL respecto al valor basal estándar; RM4,5 = reducción logarítmica ≥ 4,5 de transcritos de BCR-ABL respecto al valor basal estándar. a Incluye a los pacientes (N) con una evaluación basal válida. Para ser considerado un respondedor, el paciente debe haber logrado una mejor respuesta respecto al valor basal. Criterios aplicados a la respuesta molecular: RMM, RM4 y RM4,5 se definieron como ≤ 0,1%, ≤ 0,01% y ≤ 0,0032% del cociente BCR-ABL/ABL en la escala internacional, respectivamente (correspondientes a una reducción logarítmica ≥ 3, ≥ 4 y ≥ 4,5 del valor basal estándar) con un mínimo de 10.000, 10.000 y 32.000 transcritos de ABL evaluados por el laboratorio central, respectivamente. | ||||
En los pacientes en FC, no hubo progresiones durante el tratamiento a LMC en FA o FB.
Pacientes con LMC en FA
En pacientes con LMC Ph+ en FA, la mediana de la duración del tratamiento fue de 22,1 meses (intervalo: de 1,6 a 50,1 meses), la RHG acumulada confirmada al año (52 semanas) fue del 75,0% (IC del 95%: 19,4; 99,4), al igual que la tasa acumulada de RCyC, los 3 pacientes mantuvieron la RCyC durante el tratamiento.
Respuesta por mutaciones BCR-ABL basales
Diez pacientes de la cohorte FC presentaron mutaciones basales (A365V, E453K, E255K, E255V, Q252H, L298V [n = 1 cada una], Y253F y G250E [n = 2 cada una]). Un paciente de la cohorte FC presentó una mutación F359I que fue identificada el día 8 del estudio. Un paciente de la cohorte FA presentó 2 mutaciones (F311L y L387F) basales. En la cohorte FC, entre los pacientes con mutaciones, se observaron respuestas moleculares en 4/11 (36,4%) pacientes, 1 paciente con una mutación E255V logró una RMM y 3 pacientes con F359I, Y253F y A365V, respectivamente lograron una RM4,5. El paciente con mutaciones en la cohorte FA no logró ninguna respuesta.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Bosulif en uno o más grupos de la población pediátrica en LMC (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 - Propiedades farmacocinéticas de BOSULIF 400 mg Comp. recub. con película
Absorción
Tras la administración de una dosis única de bosutinib (500 mg) con alimentos en sujetos sanos, la biodisponibilidad absoluta fue del 34%. La absorción fue relativamente lenta, alcanzando una mediana de tiempo hasta la concentración máxima (tmax) de 6 horas. Bosutinib muestra aumentos proporcionales a la dosis en los valores de AUC y de Cmax, a lo largo del intervalo de dosis comprendido entre 200 y 600 mg. La ingesta de alimentos multiplicó por 1,8 la Cmax de bosutinib y por 1,7 el AUC de bosutinib, en relación a la administración en ayunas. En pacientes con LMC en estado estacionario, la Cmax (media geométrica, coeficiente de variación [CV]%) fue de 145 (14) ng/ml y el AUCee (media geométrica, CV%) fue de 2 700 (16) ng•h/ml tras la administración diaria de 400 mg de bosutinib con alimentos. Tras la administración diaria de 500 mg de bosutinib con alimentos, la Cmax fue de 200 (6) ng/ml y el AUCee fue de 3 640 (12) ng•h/ml. La solubilidad de bosutinib es dependiente del pH y la absorción se reduce cuando se incrementa el pH gástrico (ver sección 4.5).
Distribución
Tras la administración de una dosis única intravenosa de 120 mg de bosutinib a sujetos sanos, bosutinib tuvo un volumen medio (% coeficiente de variación [CV]) de distribución de 2 331 (32) litros, hecho que sugiere que bosutinib se distribuye de manera muy amplia por los tejidos extravasculares.
La unión de bosutinib a proteínas plasmáticas humanas fue muy alta tanto in vitro (94%) como ex vivo en sujetos sanos (96%), y dicha unión no dependió de la concentración.
Biotransformación
De los estudios realizados in vitro e in vivo se desprende que, en los seres humanos, bosutinib (compuesto original) sufre una metabolización predominantemente hepática. Se observó que, tras la administración de una dosis única o de varias dosis de bosutinib (400 o 500 mg) a humanos, los metabolitos circulantes principales fueron bosutinib oxidesclorado (M2) y bosutinib N-desmetilado (M5), y como metabolito circulante secundario, apareció N-óxido de bosutinib (M6). La exposición sistémica al metabolito N-desmetilado se cifró en un 25% respecto del compuesto inalterado, mientras que la del metabolito oxideclorado fue un 19% respecto del compuesto inalterado. Los 3 metabolitos mostraron una actividad inferior o igual al 5% de la de bosutinib en un estudio de proliferación independiente de anclaje en fibroblastos transformados con Src. En heces, los principales compuestos relacionados con el medicamento fueron bosutinib y bosutinib N-desmetilado. De los estudios realizados in vitro con microsomas hepáticos humanos, se desprende que la principal isoenzima del citocromo P450 que interviene en el metabolismo de bosutinib es CYP3A4, y en los estudios de interacción de medicamentos se ha observado que ketoconazol y rifampicina tuvieron un marcado efecto sobre la farmacocinética de bosutinib (ver sección 4.5). No se observó metabolismo de bosutinib con los CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ni 3A5.
Eliminación
En sujetos sanos a los que se les administró una dosis única intravenosa de 120 mg de bosutinib, la semivida de eliminación media (% CV) fue de 35,5 (24) horas, y el aclaramiento medio (% CV) fue de 61,9 (26) l/h. En un estudio de balance de masas con bosutinib oral, en nueve días se recuperó una media del 94,6% de la dosis total; la vía de excreción principal fueron las heces (91,3%), y en la orina se recuperó un 3,29% de la dosis. Un setenta y cinco por ciento de la dosis fue recuperado en un plazo de 96 horas. La excreción en la orina de bosutinib inalterado fue baja con aproximadamente un 1% de la dosis tanto en los sujetos sanos como en quienes tenían tumores sólidos malignos avanzados.
Poblaciones especiales
Insuficiencia hepática
Se evaluó una dosis de 200 mg de bosutinib administrada junto con alimentos en una cohorte de 18 sujetos con insuficiencia hepática (de clases A, B y C de Child-Pugh) y en 9 sujetos sanos. En las clases A, B y C de Child-Pugh, la Cmax de bosutinib en plasma se multiplicó por 2,4; 2 y 1,5 veces, respectivamente; y el AUC de bosutinib en plasma se multiplicó por 2,3; 2 y 1,9 veces, respectivamente. La t½ de bosutinib aumentó en los pacientes con insuficiencia hepática en comparación con los sujetos sanos.
Insuficiencia renal
En un estudio sobre la insuficiencia renal se administraron 200 mg de bosutinib con alimento a 26 sujetos con insuficiencia renal leve, moderada o grave y a 8 voluntarios sanos. La insuficiencia renal se basaba en un ClCr (calculado según la fórmula de Cockcroft-Gault) de < 30 ml/min (insuficiencia renal grave), 30 ≤ ClCr ≤ 50 ml/min (insuficiencia renal moderada) o 50 < ClCr ≤ 80 ml/min (insuficiencia renal leve). Los sujetos con insuficiencia renal moderada y grave experimentaron un aumento del AUC con respecto a los voluntarios sanos del 35% frente al 60%. La exposición máxima Cmax aumentó un 28% y un 34% en los grupos moderados y graves, respectivamente. La exposición a bosutinib no aumentó en sujetos con insuficiencia renal leve. La semivida de eliminación de bosutinib en sujetos con insuficiencia renal fue similar a la de los sujetos sanos.
Los ajustes de dosis para la insuficiencia renal se basaron en los resultados de este estudio y en la farmacocinética lineal conocida de bosutinib en el rango de dosis de 200 a 600 mg.
Edad, sexo y raza
No se han llevado a cabo estudios formales para evaluar los efectos de estos factores demográficos. De los análisis de farmacocinética en la población llevados a cabo en pacientes con leucemia Ph+ o con tumores sólidos malignos y en sujetos sanos, se desprende que la edad, el sexo o el peso corporal no tienen efectos relevantes desde el punto de vista clínico. Los análisis farmacocinéticos poblacionales revelaron que los asiáticos tenían un aclaramiento reducido en un 18% que corresponde a un aumento de aproximadamente el 25% en la exposición a bosutinib (AUC).
Población pediátrica
Aún no se ha estudiado Bosulif en niños y adolescentes menores de 18 años de edad.
5.3 - Datos preclínicos sobre seguridad de BOSULIF 400 mg Comp. recub. con película
Se ha evaluado bosutinib en estudios de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, toxicidad para la reproducción y fototoxicidad.
Farmacología de seguridad
Bosutinib no ejerció efecto alguno sobre la función respiratoria. En un estudio referido al sistema nervioso central (SNC) efectuado con ratas tratadas con bosutinib, éstas mostraron una disminución del diámetro pupilar y alteraciones de la marcha. No se determinó un nivel sin efecto observado (NOEL, por sus siglas en inglés) correspondiente al diámetro pupilar, pero el NOEL correspondiente a las alteraciones de la marcha ocurrió a exposiciones de, aproximadamente, 11 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg y 8 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 500 mg (teniendo en cuenta la Cmax de la fracción libre en las respectivas especies). De la actividad in vitro de bosutinib que se observó en estudios realizados acerca de hERG (conducción nerviosa) parece desprenderse que bosutinib puede prolongar la repolarización ventricular cardiaca (QTc). En un estudio de bosutinib por vía oral en perros, bosutinib no produjo cambios en la presión arterial, ni arritmias auriculares o ventriculares anómalas, ni prolongación de PR, QRS o QTc del ECG a exposiciones de hasta 3 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg y 2 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 500 mg (teniendo en cuenta la Cmax de la fracción libre en las respectivas especies). Sí que se observó un aumento retrasado de la frecuencia cardiaca. En un estudio por vía intravenosa en perros, se observaron aumentos transitorios de la frecuencia cardiaca, disminuciones de la presión arterial y una prolongación mínima del QTc (< 10 mseg) a exposiciones comprendidas aproximadamente entre 6 y 20 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg y entre 4 y 15 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 500 mg (teniendo en cuenta la Cmax de la fracción libre en las respectivas especies). No fue posible llegar a conclusiones sobre la relación que podía existir entre los efectos observados y el tratamiento con el medicamento.
Toxicidad a dosis repetidas
Se llevaron a cabo estudios sobre toxicidad a dosis repetidas, de hasta 6 meses de duración en ratas y de hasta 9 meses de duración en perros, de los que se desprende que el principal sistema afectado por la toxicidad de bosutinib es el aparato digestivo. Los signos clínicos de toxicidad incluyeron cambios en las heces, que se asociaron a una disminución de la ingesta de alimentos, así como pérdida de peso corporal que, en ocasiones, condujo a la muerte o a la eutanasia.
Histopatológicamente se observó dilatación de la luz intestinal, hiperplasia de células caliciformes, hemorragias, erosión y edema del tracto intestinal, así como eritrocitosis sinusoidal y hemorragias en los ganglios linfáticos mesentéricos. El hígado también ha sido identificado en ratas como órgano afectado por la toxicidad. Las toxicidades se caracterizaron por un aumento en el peso del hígado en correlación con una hipertrofia hepatocelular, que tuvo lugar en ausencia de enzimas hepáticas elevadas o signos microscópicos de citotoxicidad hepatocelular, y cuya relevancia en el hombre es desconocida. De la comparación entre las exposiciones en especies diferentes se desprende que las exposiciones que no produjeron acontecimientos adversos en los estudios de toxicidad a 6 y 9 meses en ratas y perros, respectivamente, fueron similares a la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg o 500 mg (teniendo en cuenta el AUC de bosutinib libre en las respectivas especies).
Genotoxicidad
En los estudios de genotoxicidad en sistemas bacterianos in vitro y en sistemas mamíferos in vitro e in vivo, con y sin activación metabólica, no se hallaron evidencias de potencial mutagénico de bosutinib.
Toxicidad para la reproducción y el desarrollo
En un estudio de fertilidad en ratas, se observó una ligera disminución de la fertilidad en ratas machos. En ratas hembras se observó un aumento de las reabsorciones de embriones, así como reducciones del número de implantaciones y de embriones viables. La dosis a la que no se observó ningún efecto adverso sobre la reproducción en las ratas machos (30 mg/kg/día) y hembras (3 mg/kg/día) corresponde a exposiciones equivalentes a 0,6 y 0,3 veces, respectivamente, la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg y 0,5 veces y 0,2 veces respectivamente, la exposición en seres humanos a la dosis clínica de 500 mg (teniendo en cuenta el AUC desligado en las respectivas especies). No se puede excluir un efecto sobre la fertilidad masculina (ver sección 4.6).
En un estudio realizado sobre la transferencia a través de la placenta en ratas Sprague-Dawley preñadas, se evaluó la exposición fetal a la radioactividad vinculada al bosutinib durante el embarazo. En un estudio de desarrollo pre– y posnatal en ratas, con dosis ≥ 30 mg/kg/día hubo una reducción en el número de crías nacidas, y con dosis de 70 mg/kg/día se produjo un aumento de la incidencia de la pérdida total de la camada y una reducción en el crecimiento de las crías tras el nacimiento. La dosis a la que no se observó ningún efecto adverso sobre el desarrollo (10 mg/kg/día) dio como resultado exposiciones equivalentes a 1,3 veces y 1,0 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg y de 500 mg, respectivamente (teniendo en cuenta el AUC de bosutinib libre en las respectivas especies). En un estudio de toxicidad para el desarrollo en conejos con dosis tóxicas en las madres, se observaron anomalías fetales (fusión del esternón, y 2 fetos presentaron diversas anomalías viscerales), así como una ligera reducción del peso corporal de los fetos. La exposición a la dosis más alta probada en conejas (10 mg/kg/día) que no provocó efectos adversos en los fetos fue 0,9 veces y 0,7 veces la exposición en los seres humanos resultante de la administración de la dosis clínica de 400 mg o de 500 mg, respectivamente (teniendo en cuenta el AUC de bosutinib libre en las respectivas especies).
Tras una única administración por vía oral (10 mg/kg) de bosutinib radiomarcado con [14C] a ratas Sprague-Dawley lactantes, la radioactividad fue excretada rápidamente en la leche materna, incluso en 0,5 horas tras la administración. La concentración de la radioactividad en la leche fue hasta 8 veces más alta que en el plasma. Es por ello que en el plasma de las crías lactantes aparecieron concentraciones apreciables de radioactividad.
Carcinogenicidad
Bosutinib no fue carcinogénico en los estudios de potencial carcinogénico a 2 años en ratas y a 6 meses en ratones rasH2.
Fototoxicidad
Se ha demostrado que bosutinib tiene capacidad para absorber luz en las franjas UV-B y UV-A del espectro y que se difunde dentro la piel y de la úvea de las ratas pigmentadas. Sin embargo, bosutinib no mostró potencial fototóxico para la piel ni para los ojos en ratas pigmentadas expuestas a bosutinib en presencia de radiación UV a exposiciones a bosutinib hasta 3 y 2 veces la exposición en seres humanos resultante de la administración de la dosis clínica de 400 mg o 500 mg, respectivamente (teniendo en cuenta la Cmax de la fracción libre en las respectivas especies).
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de BOSULIF 400 mg Comp. recub. con película
Núcleo del comprimido
Celulosa microcristalina (E460)
Croscarmelosa de sodio (E468)
Poloxámero 188
Povidona (E1201)
Estearato de magnesio (E470b)
Recubrimiento con película
Bosulif 100 mg comprimidos recubiertos con película
Alcohol polivinílico
Dióxido de titanio (E171)
Macrogol 3350
Talco (E553b)
Óxido de hierro amarillo (E172)
Bosulif 400 mg comprimidos recubiertos con película
Alcohol polivinílico
Dióxido de titanio (E171)
Macrogol 3350
Talco (E553b)
Óxido de hierro amarillo (E172)
Óxido de hierro rojo (E172)
Bosulif 500 mg comprimidos recubiertos con película
Alcohol polivinílico
Dióxido de titanio (E171)
Macrogol 3350
Talco (E553b)
Óxido de hierro rojo (E172)
6.2 - Incompatibilidades de BOSULIF 400 mg Comp. recub. con película
No procede.
6.3 - Período de validez de BOSULIF 400 mg Comp. recub. con película
4 años.
6.4 - Precauciones especiales de conservación de BOSULIF 400 mg Comp. recub. con película
No requiere condiciones especiales de conservación.
6.5 - Naturaleza y contenido del recipiente de BOSULIF 400 mg Comp. recub. con película
Blíster blanco opaco de tres capas de PVC/Policlorotrifluoroetileno/PVC sellado con una lámina de aluminio que se rompe al presionarla, conteniendo 14 o 15 comprimidos.
Bosulif 100 mg comprimidos recubiertos con película
Cada estuche contiene 28, 30 o 112 comprimidos.
Bosulif 400 mg comprimidos recubiertos con película
Cada estuche contiene 28 o 30 comprimidos.
Bosulif 500 mg comprimidos recubiertos con película
Cada estuche contiene 28 o 30 comprimidos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de BOSULIF 400 mg Comp. recub. con película
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Bruxelles
Bélgica
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bosulif 100 mg comprimidos recubiertos con película
EU/1/13/818/001
EU/1/13/818/002
EU/1/13/818/005
Bosulif 400 mg comprimidos recubiertos con película
EU/1/13/818/006
EU/1/13/818/007
Bosulif 500 mg comprimidos recubiertos con película
EU/1/13/818/003
EU/1/13/818/004
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 27/marzo/2013
Fecha de la última renovación: 31/marzo/2022
10. - FECHA DE LA REVISIÓN DEL TEXTO
04/2023
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
PRESENTACIONES Y PRECIO
BOSULIF 100 mg, 28 comprimidos recubiertos con película: PVL: 721,65 €; PVP: 777,56€; PVP IVA: 808,66€
BOSULIF 400 mg, 28 comprimidos recubiertos con película: PVL: 2.886,59 €; PVP: 2.942,50 €; PVP IVA: 3.060,20 €
BOSULIF 500 mg, 28 comprimidos recubiertos con película: PVL: 3.608,23 €; PVP: 3.664,14 €; PVP IVA: 3.810,71€
CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN
Con receta médica. Uso Hospitalario.
CONDICIONES DE LA PRESTACIÓN FARMACÉUTICA
Financiado por el Sistema Nacional de Salud, sin aportación.
Para información adicional, por favor, contacte con el Centro de Información Médico-Farmacéutica llamando al + 34 914909900 o consulte nuestra página web www.pfizer.es.
Nota relativa a la indicación de leucemia mieloide crónica con cromosoma Ph+ en fase crónica recién diagnosticada:
La indicación terapéutica relativa al tratamiento de pacientes adultos con leucemia mieloide crónica con cromosoma Philadelphia positivo (LMC Ph+) en fase crónica (FC) recién diagnosticada no se encuentra incluida en la prestación farmacéutica del SNS.

