Informe de Posicionamiento Terapéutico de elvitegravir/cobicistat/ emtricitabina/tenofovir alafenamida (Genvoya®) en infección por VIH
Informe de Posicionamiento Terapéutico de elvitegravir/cobicistat/ emtricitabina/tenofovir alafenamida (Genvoya®) en infección por VIH
Fecha de publicación: 31 de mayo de 2016
Desde el comienzo de la epidemia del virus de la inmunodeficiencia humana (VIH), aproximadamente 78 millones de personas han sido infectadas. Alrededor de 35 millones de personas en todo el mundo viven con el VIH. La infección por el VIH sigue siendo una enfermedad que amenaza la vida de aquellas personas infectadas que no reciben un tratamiento óptimo iniciado lo suficientemente temprano y/o se infectaron con variantes del virus que son resistentes a varias clases de medicamentos antirretrovirales (1).
Las estrategias terapéuticas para el tratamiento de la enfermedad del VIH-1 han avanzado de manera significativa por la disponibilidad de la terapia antirretroviral de alta eficacia (TAR). La introducción de esta terapia se ha asociado a una disminución drástica en la morbimortalidad relacionada con el sida (2). El objetivo de la terapia antirretroviral es evitar la progresión de la enfermedad, mejorar la calidad de vida, restaurar y preservar la función del sistema inmunológico, suprimir la replicación del VIH-1, y mantenerla suprimida, y prevenir la transmisión del virus.
Las directrices actuales de tratamiento para pacientes sin TAR previo o naïve, recomiendan un abordaje terapéutico basado en combinaciones de tres fármacos y, además, recomiendan valorar la indicación de inicio de tratamiento a todo paciente infectado con el VIH-1 independientemente de los niveles de CD4. Este esquema terapéutico debe incluir dos inhibidores de la transcriptasa inversa análogos de nucleósido o nucleótido (ITIAN) y un tercer fármaco inhibidor de la integrasa (InInt), inhibidor de la transcriptasa inversa no nucleósido (ITINN) o un inhibidor de la proteasa potenciado (IP/p) (3).
A la hora de seleccionar una u otra familia de antirretrovirales, es importante hacer una valoración individualizada en el momento de inicio del TAR y de los fármacos que deben formar parte del régimen inicial, sopesando las ventajas e inconvenientes de cada una de las opciones, adaptando el esquema terapéutico al estilo de vida, comorbilidades, posibles interacciones y valorando el riesgo de mala adherencia.
Las distintas guías hacen hincapié en la influencia de ciertos factores en el balance riesgo/beneficio del TAR como la toxicidad a medio-largo plazo de los antirretrovirales, los problemas de adherencia, la aparición de resistencias, las interacciones medicamentosas y el impacto en la calidad de vida (4). Debido a que el número de antirretrovirales (AR) ha aumentado, lo que permite nuevas combinaciones, se ha mejorado la eficacia y seguridad y ahora existe la posibilidad de simplificar el TAR.
Las ventajas de los regímenes basados en InInt incluyen una disminución rápida (4-8 semanas) de la carga viral plasmática (CVP) y que raramente se transmiten resistencias a InInt, por lo que tienen su lugar en la infección aguda y la disminución detransmisión del virus. Sin embargo, InInt como elvitegravir necesita potenciación farmacológica porque es intensamente metabolizado por las enzimas del citocromo P4503A (CYP3A) y necesita ser potenciado con cobicistat que es un inhibidor de esta enzima oxidativa.
Los progresos en el tratamiento antirretroviral no solo incluyen avances en la eficacia de los fármacos para controlar la enfermedad, sino que a lo largo de los años, se han conseguido fármacos menos tóxicos y con mayor tolerabilidad. Actualmente, además, existen combinaciones a dosis fijas en un único comprimido. Esto simplifica considerablemente el régimen AR, es decir, con una menor carga de comprimidos y menor frecuencia, se logra una supresión virológica sostenible.
ELVITEGRAVIR/COBICISTAT/EMTRICITABINA/TENOFOVIR ALAFENAMIDA(GENVOYA®) (5,6)
Genvoya® es un medicamento que combina a dosis fijas elvitegravir (EVG) 150 mg, cobicistat (COBI) 150 mg, emtricitabina (FTC) 200 mg y tenofovir alafenamida (TAF) 10 mg. Está indicado para el tratamiento de adultos y adolescentes (de 12 años de edad o mayores con un peso corporal de al menos 35 kg) infectados con el virus VIH-1 sin ninguna mutación conocida asociada con resistencia a los inhibidores de la integrasa, emtricitabina o tenofovir.
La posología recomendada es un comprimido una vez al día, junto con comida, evitando masticar o dividir los comprimidos.
Farmacología
Farmacodinamia
Elvitegravir (EVG)es un inhibidor de la integrasa del VIH-1, enzima codificada por el VIH-1. La inhibición impide la integración del ADN del VIH-1 en el ADN genómico del huésped.
Cobicistat (COBI)es un inhibidor selectivo de las enzimas del citocromo CYP3A, potenciando la exposición sistémica a los sustratos del CYP3A (como EVG). Es un análogo estructural de ritonavir, aunque a diferencia de éste último no presenta actividad antirretroviral.
Emtricitabina (FTC)es un inhibidor de la transcriptasa inversa análogo de nucleósido (ITIAN). Una vez fosforilado en el interior de la célula, se incorpora al ADN viral mediante la transcriptasa inversa (TI) e inhibe la replicación del VIH interrumpiendo la cadena del ADN.
Tenofovir alafenamida fumarato (TAF)es un profármaco del tenofovir, inhibidor de la transcriptasa inversa análogo de nucleótido (ITIAN). Una vez difosforilado se incorpora al ADN viral mediante la TI e interrumpe la cadena de ADN.
Farmacocinética
La administración de Genvoya® con una comida ligera o con una comida grasa no afectó a los valores globales de exposición a tenofovir alafenamida en un grado clínicamente relevante.
En ensayos in vivo, TAF se hidroliza en las células para formar tenofovir (metabolito principal), que es fosforilado al metabolito activo tenofovir difosfato. En los ensayos clínicos en humanos, una dosis oral de 10 mg de TAF en Genvoya® dio lugar a unas concentraciones intracelulares de tenofovir difosfato más de 4 veces superiores y una concentración 90 % menor en plasma en comparación con una dosis oral de 245 mg de tenofovir disoproxil administrado en elvitegravir/cobiscistat/emtricitabina/tenofovir disoproxil (Stribild®; E/C/F/TDF).
El análisis de los datos farmacocinéticos del estudio GS-US-292-0106 en pacientes adolescentes naïve sugiere que las exposiciones plasmáticas a TAF y tenofovir (TFV) son equivalentesen adultos y adolescentes. No hay razones para pensar que lacaptación de TAF y conversión intracelular a TFV estuviera impedida en adolescentes comparada con adultos.
La unión a proteínas plasmáticas (UPP) es alta para elvitegravir (98-99 %) y cobicistat (97-98 %). La UPP de emtricitabina y tenofovir es despreciable in vitro, pero ex vivo TAF mostró una unión del 80 %.
La eliminación de EVG/r y COBI es mayoritariamente por heces, y en menor medida, por orina. En cambio, FTC se elimina por orina y en menor medida por heces, y TFV se elimina por filtración glomerular y secreción activa en orina.
En cuanto a las posibles interacciones con otros medicamentos, cabe esperar que la administración concomitante de Genvoya® con fármacos que sean inhibidores del citocromo P450 3A (CYP3A) puede disminuir la eficacia el EVG y el COBI y favorecer la aparición de resistencias. Está contraindicado el uso de antagonistas de algunos receptores adrenérgicos alfa 1 (alfuzosina), antiarrítmicos (amiodarona, quinidina), antiepilépticos (carbamazepina, fenobarbital, fenitoína), antimicobacterianos (rifampicina), derivados ergóticos (dihidroergotamina, ergometrina, ergotamina), fármacos estimulantes de la motilidad gastrointestinal (cisaprida), medicamentos a base de plantas (Hypericum perforatum), inhibidores de la HMG Co-A reductasa (lovastatina, simvastatina), neurolépticos (pimozida), inhibidores de la PDE-5 (sildenafilo para el tratamiento de la hipertensión arterial pulmonar) y sedantes/hipnóticos (midazolam administrado por vía oral, triazolam ).
No hay ensayos adecuados y bien controlados de Genvoya® o de sus componentes en mujeres embarazadas.
La justificación de la elección de dosis de 10 mg de TAF se basa en estudios farmacocinéticos, como el GS-US-292-0103, en los que se observó que el efecto de TAF 10 mg asociado a un inhibidor enzimático como COBI presentaba el mismo efecto que el TAF 25 mg en monoterapia.
Eficacia
La eficacia clínica de este fármaco se basa en un primer estudio fase II que se realizó para avalar la dosis de 10 mg de TAF y evaluar la eficacia y en tres estudios principales de fase III. Dos de los tres pivotales se llevaron a cabo en pacientes naïve adultos para demostrar la no inferioridad de elvitegravir/cobiscistat/ emtricitabina/ tenofovir alafenamida (E/C/F/TAF) frente a E/C/F/TDF, y el tercero en pretratados para evaluar la eficacia y seguridad del cambio a E/C/F/TAF en pacientes con respuesta virológica suprimida. Además, se han presentado 2 estudios abiertos de soporte en pacientes naïve adolescentes y en pacientes con insuficiencia renal.
Estudio dosis-respuesta
El GS-US-292-0102 fue un estudio fase II, aleatorizado, doble ciego, multicéntrico, con control activo, en pacientes naïve para evaluar la seguridad y la eficacia de E/C/F/TAF frente a E/C/F/TDF. Tras la semana 48, finalizó el doble ciego y se inició una fase abierta que permitió el cruce de pacientes de E/C/F/TDF a E/C/F/TAF y, además, permitió la adhesión de pacientes con carga viral suprimida que habían recibido regimenes con darunavir (DRV) más cobicistat en el estudio GS-US-299-0102.
El objetivo principal fue evaluar la eficacia de E/C/F/TAF frente a E/C/F/TDF, medida como respuesta virológica (carga viral inferior a 50 copias/mL) en la semana 48.
Se incluyeron 170 pacientes aleatorizados (2:1) a E/C/F/TAF y E/C/F/TDF, respectivamente. No se observaron diferencias en la respuesta virológica en la semana 48 (88,4 % E/C/F/TAF vs 87,9 % E/C/F/TDF; p=0,84). Se mantuvieron altos porcentajes de respuesta virológica hasta la semana 96 y no hubo casos de repunte virológico.
En la fase abierta, 105 pacientes continuaron con E/C/F/TAF, y161 cambiaron a E/C/F/TAF desde E/C/F/TDF (n=53), DRV/C/F/TAF (n=70) y DRV/C/F/TDF (n=38). Se observó que los pacientes que habían sido aleatorizados desde el inicio a E/C/F/TAF mantenían en un 84,8 % la carga viral suprimida en la semana 144. En aquellos que se habían cambiado de E/C/F/TDF a E/C/F/TAF se mantenían en un 98,6 % y 100 % en la semana 48.
Estudios pivotales en pacientes naïve
GS-US-292-0104 y GS-US-292-0111 fueron estudios de fase III, aleatorios, doble ciego, con control activo en pacientes naïve adultos VIH-1 positivos en los que se comparó la eficacia, seguridad y farmacocinética de E/C/F/TAF frente a E/C/F/TDF (7). Los dos estudios tuvieron el mismo diseño, resumido en la Tabla 1.
Tabla 1: Diseño de los estudios pivotales (GS-US-292-0104, GS- US-292-0111)
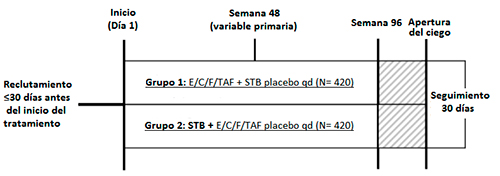
El objetivo principal fue la respuesta virológica en la semana 48. La variable primaria fue evaluar la eficacia de E/C/F/TAF frente a E/C/F/TDF, medida como la proporción de sujetos en la semana 48 con niveles de carga viral (copias de RNA VIH-1, a partir de ahora CV) inferiores a 50 copias/mL definidos por el algoritmo snapshot de la FDA. Se definió fracaso virológico si se detectaban valores mayores 50 copias/mL en la semana 48, si se había abandonado el estudio antes de la semana 48 por falta de eficacia o bien, si se había discontinuado por otras causas y la última CV disponible era superior a 50 copias/mL. Se categorizó como respuesta subóptima cuando no se producía una disminución de al menos 1 log10en la semana 8 respecto a la visita basal. Los objetivos secundarios incluían el porcentaje de pacientes con CV inferior a 20 copias/mL, cambios en el recuento de CD4, ambos a las semanas 48 y 96, y objetivos de seguridad relacionados con la reducción de la densidad mineral ósea y riesgo de fractura y la nefrotoxicidad.
En el análisis primario de los resultados se incluyó la población por intención de tratar (ITT) y en el análisis secundario la población por protocolo (PP).
Para la comparación de ambos grupos de tratamiento se realizó un estudio de no inferioridad. Se definió que E/C/F/TAF era no- inferior a E/C/F/TDF si el límite inferior del IC95 % de la diferencia en la respuesta entre ambos grupos de tratamiento era mayor a -12% en la semana 48. La aleatorización se realizó estratificando por carga viral, recuento de CD4 y región geográfica de procedencia.
De los 2175 pacientes inicialmente aleatorizados, 1733 recibieron al menos una dosis de tratamiento: 833 recibieron TAF y867 recibieron TDF. Un 89 % de los pacientes con TAF y un 87 % de los pacientes con TDF continuaban en la semana 96. Las características demográficas fueron similares en los dos grupos. La media de edad fue de 36 años (18-76), el 85 % eran hombres y el 57% de raza blanca. La mediana de copias/ml al inicio del estudio fue4,58 log10copias/mL (17,4 % con > 100.000 copias/mL y 5,2 % con> 400.000 copias/mL). Al inicio un 2,9 % tenía un recuento de CD4< 50 células/µL un 10,3 % tenía 50 a 200 células/µL y un 31,5 >500 células/µL. El valor de aclaramiento de creatinina (ClCr) al inicioera 115,6 mL/min.
E/C/F/TAF cumplió los criterios de no inferioridad en cuanto a la consecución de un ARN del VIH-1 < 50 copias/ml al compararlo con E/C/F/TDF. Los resultados combinados de ambos estudios a las48 y a las 96 semanas se muestran en la tabla 2.
El análisis de subgrupos no mostró diferencias significativas.
Tabla 2. Resultados virológicos combinados de los ensayos GS- US -292-0104 y GS-US-292-0111 en las semanas 48 y 96a,b
| Semana 48 | Semana 96 | |||
| Genvoya (n = 866) | E/C/F/TDF (n = 867) | Genvoya (n = 866) | E/C/F/TDF (n = 867) | |
| ARN del VIH-1 < 50 copias/ml | 92% | 90% | 87% | 85% |
| Diferencia entre tratamientos | 2,0% (IC del 95%: -0,7% a 4,7%) | 1,5% (IC del 95%: -1,8% a 4,8%) | ||
| ARN del VIH-1 ≥ 50 copias/mlcc | 4% | 4% | 5% | 4% |
| Ausencia de datos virológicos en la ventana de la semana 48 o 96 | 4% | 6% | 9% | 11% |
| Proporción (%) de pacientes con ARN del VIH-1 < 50 copias/ml por subgrupo | ||||
| Carga viral basal ≤ 100.000 copias/ml > 100.000 copias/ml | 629/670 (94%) 171/196 (87%) | 610/672 (91%) 174/195 (89%) | 587/670 (88%) 163/196 (83%) | 573/672 (85%) 166/195 (85%) |
| Recuento basal de células CD4+ < 200 células/mm3 ≥ 200 células/mm3 | 96/112 (86%) 703/753 (93%) | 104/117 (89%) 680/750 (91%) | 93/112 (83%) 657/753 (87%) | 97/117 (83%) 642/750 (86%) |
| ARN del VIH-1 < 20 copias/ml | 84,4% | 84,0% | 81,5% | 80,2% |
| Diferencia entre tratamientos | 0,4% (IC del 95%: -3,0% a 3,8%) | 1,5% (IC del 95%: -2,2% a 5,2%) | ||
E/C/F/TDF = elvitegravir/cobicistat/emtricitabina/ tenofovir disoproxil fumarato
a La ventana de la semana 48 fue entre los días 294 y 377 (ambos inclusive); la ventana de la semana 96 fue entre los días 630 y 713 (ambos inclusive).
b En ambos ensayos se estratificó a los pacientes según su ARN del VIH-1 basal (≤100.000 copias/ml, > 100.000 copias/ml a ≤ 400.000 copias/ml o > 400.000 copias/ml), según el recuento de células CD4+ (< 50 células/μl, 50-199 células/μl o ≥ 200 células/μl) y según la región (EEUU o fuera de EEUU).
c Incluye a los pacientes que tenían ≥ 50 copias/ml en la ventana de la semana 48 o 96, los pacientes que abandonaron precozmente debido a ausencia o pérdida de la eficacia, los pacientes que abandonaron por motivos distintos de un acontecimiento adverso (AA), muerte o ausencia o pérdida de la eficacia y que en el momento del abandonotenían un valor viral ≥ 50 copias/ml.
d Incluye a los pacientes que abandonaron debido a un AA o a la muerte en cualquier momento desde el día 1 hasta la ventana temporal, si esto dio lugar a una ausencia de datos virológicos sobre el tratamiento durante la ventana especificada.
e Incluye a los pacientes que abandonaron por motivos distintos de un AA, muerte o ausencia o pérdida de la eficacia, p. ej., que retiraron el consentimiento, se perdieron para el seguimiento, etc.
Estudio del cambio de tratamiento con carga viral suprimida
GS-US-292-0109 fue un estudio fase III, abierto en el que se evaluó el cambio de regímenes que contenían TDF a la combinación a dosis fijas que contienen TAF (E/C/F/TAF) (8).
Se incluyeron pacientes mayores de 18 años, que se mantenían con CV indetectable durante al menos 6 meses con algunos de los siguientes regímenes de tratamiento: E/C/F/TDF, efavirenz/ emtricitabina/tenofovir disoproxil (EFV/FTC/TDF), atazanavir potenciado con ritonavir (ATV/r) más FTC/TDF o ATV/COBI más FTC/TDF.
Los pacientes se aleatorizaron 2:1, estratificando por el régimen anterior, al grupo 1, E/C/F/TAF, y al grupo 2, FTC/TDF más tercer agente.
El objetivo principal fue evaluar la no inferioridad del cambio de tratamiento en pacientes con carga viral suprimida (CV inferior a50 copias/mL). Las variables primarias y secundarias fueron lasmismas que en los estudios pivotales en pacientes naïve, así como el margen de -12 % de no inferioridad.
Los resultados demostraron la no inferioridad del cambio aE/C/F/TAF desde regímenes con FTC/TDF más tercer agente (tabla3), siendo la diferencia estadísticamente significativa para superioridad en el grupo de pacientes que cambiaron a Genvoya®.
No hubo diferencias significativas entre los subgrupos.
Tabla 3. Respuesta virológica en la semana 48 GS-US-292-0109
| E/C/F/TAF n ( %) | FTC/TDF + 3er agente n ( %) | Diferenciaa (IC95 %) p-valueb | |
| N=959 | N=477 | ||
| Respuesta virológica (CV < 50 copias/mL) | 932 (97,2 %) | 444 (93,1 %) | 4,1 % (1,6 a 6,7 %) P<0,001 |
| Fracaso virológico (CV ≥ 50 copias/mL) | 10 (1 %) | 6 (1,3 %) |
a. La diferencia de los porcentajes de respuesta virológica entre los tratamientos y su IC95 % se calcularon en base a las proporciones MH ajustadas por tratamiento anterior b. P-value para el test de superioridad comparando los porcentajes de respuesta virológica a partir del test estratificado CMH por tratamiento anterior.
Poblaciones especiales
El GS-US-292-0112 fue un estudio de fase III, abierto, de seguridad del E/C/F/TAF a dosis fijas en pacientes con VIH-1 positivo con insuficiencia renal leve o moderada (9).
Fue diseñado para evaluar el perfil de seguridad de E/C/F/TAF en pacientes con insuficiencia renal estable (ClCrCG30-69 mL/min) en la semana 24. La variable principal fue el cambio en la tasa de filtarción glomerular. La eficacia se midió con las mismas variables que los estudios descritos anteriormente. La inclusión de pacientes se realizó en dos cohortes. La cohorte 1 contenía 242 pacientes que cambiaron a E/C/F/TAF desde regímenes con los que habían conseguido CV indetectable durante al menos 6 meses (conteniendo o no TDF). Y la cohorte 2, contenía a 6 pacientes naïve que tenían al menos ≥1.000 copias/mL.
El 97,5 % tenían CV < 50 copias/mL y el 2,5 % ≥ 50 y ≤100.000 copias/mL. El 11,6 % estaban sintomáticos y el 14 %estaban diagnosticados de SIDA.
La mediana de ClCrCG al inicio fue 55,6 mL/min. Del total, un33,1 % tenían un ClCrCG < 50 mL/min, el 63,6 % tenía un ClCrCG entre 30-59 mL/min, el 42,3 % tenía proteinuria significativa (relación proteína/creatinina > 200 mg/g) y el 48,9 % teníaalbuminuria clínicamente significativa (relación proteína/creatinina≥ 30 mg/g). El 65 % de pacientes de la cohorte 1 habían recibido regímenes con TDF, de los cuales un 22,3 % de la enfermedad renal crónica se asociaba al uso de TDF.
A las 48 semanas de tratamiento no hubo cambios clínicamente apreciables en el aclaramiento de creatinina. Los resultados fueron similares en pacientes con ClCrCG < 50 o ≥ 50 mL/min o en los que habían recibido tratamientos previos con TDF
El GS-US-292-0106 fue un estudio fase II/III, abierto, de farmacocinética, seguridad (objetivos primarios) y actividad antiviral (objetivo secundario) de E/C/F/TAF a dosis fijas en pacientes naïve adolescentes VIH-1 positivos. El estudio abiertoincluyó un solo brazo con 50 pacientes de entre 12 y 18 años de edad, con peso corporal ≥ 35 Kg, CV ≥ 1.000 copias/mL, recuento de CD4 > 100 células/µL y filtrado glomerular ≥ 90 mL/min/1,73 m2. La primera parte consistió en evaluar la farmacocinética en la semana 4 con una parte de la población (25 pacientes), y la segunda parte (50 pacientes), consistió en evaluar la eficacia y seguridad enla semana 24. Las variables fueron las mismas que en los estudios anteriormente descritos.
Los resultados mostraron un porcentaje de respuesta virológica del90 % (n=45). El 8 % (n=4) se catalogó como fracaso virológico. La media (SD) de incremento en el recuento de CD4 fue 191 (175,2),menor que en los estudios de adultos en la semana 48 y con mayordesviación estándar, aunque puede deberse a que el tamaño de la muestra es considerablemente inferior.
Seguridad
Los datos de seguridad se centran en los 2.396 sujetos de los estudios de fase II y III que recibieron E/C/F/TAF.
Los efectos adversos más frecuentes (≥ 1/10) fueron diarrea, náuseas, cefalea y enfermedades del aparato respiratorio superior, muy similares al tratamiento con E/C/F/TDF.
En los estudios pivotales se describieron efectos adversos graves (grado 3 y 4) en una proporción baja en el 8,1 % en grupo E/C/F/TAF y 4,4 % en el grupo E/C/F/TDF, pero solo 2 y 3 respectivamente se asociaron al uso del medicamento. En el estudio de cambio de tratamiento con respuesta virológica suprimida (RVS) se describieron efectos adversos graves en el 4,4 % en ambos grupos, pero ninguno se asoció al fármaco en el grupo de E/C/F/TAF.
Hubo aumentos mayores significativos de colesterol total, HDL y LDL en el grupo de E/C/F/TAF pero no en el cociente colesterol total/HDL.
No se observaron cambios clínicamente relevantes en los parámetros hematológicos. Además, no hubo muertes asociadas al fármaco en ningún estudio.
Un riesgo identificado es que interrumpir el tratamiento podría empeorar los problemas hepáticos en pacientes co-infectados con VHB, debido a que FTC y TAF tienen actividad frente al VHB.
En este sentido debería tenerse en cuenta para los pacientes coinfectados con hepatitis B aunque los datos son aún escasos y a corto plazo, están disponibles recientemente los resultados a 48 semanas del estudio GS110 en hepatitis B y en switch en paciente coinfectados HIV/VHB (10, 11, 12).
Un riesgo potencial importante asociado al tratamiento antirretroviral es la ideación suicida y la tentativa de suicidio en pacientes que tienen depresión o problemas de salud mental anteriores. El plan de gestión de riesgos acordado incluye un estudio post autorización prospectivo de los casos de ideación o tentativa de suicidio. Además incluye otros estudios tales como un estudio observacional sobre información en embarazo y estudios in vitro para identificar interacciones potenciales fármaco-fármaco.
Reducción de la densidad mineral ósea
En los ensayos realizados con pacientes naïve, E/C/F/TAF se asoció con menor reducción de la densidad mineral ósea (DMO; medida mediante análisis DEXA en la cadera y en la columna lumbar) en comparación con E/C/F/TDF después de 48 semanas de tratamiento. Estos resultados se mantienen hasta la semana 96 e incluso se observa una mejoría en la DMO en columna (13). En el cambio a E/C/F/TAF desde un régimen que contiene TDF, seobservaron ligeras mejorías en la DMO en la semana 4, en comparación con el grupo de FTC/TDF+ 3eragente.
Nefrotoxicidad
En cuanto a la seguridad renal, no se describieron casos de tubulopatía proximal renal (incluido el síndrome de Fanconi) en pacientes que habían recibido E/C/F/TAF en ningún período de tiempo, con resultados hasta la semana 96. En los ensayos en pacientes naïve se observó que la Cr aumentaba al inicio del tratamiento respecto al nivel basal, pero siempre inferior significativamente al grupo de E/C/F/TDF (figura 1). Se observó que el nivel de creatinina se mantuvo en el tiempo hasta la semana96 (10); de la manera concordante, se observa que el ClCr a partir de la semana 60 se mantiene en E/C/F/TAF mientras que va disminuyendo para E/C/F/TDF. Debe tenerse en cuenta que en lostres ensayos pivotales se permitía incluir pacientes con una tasa de filtración glomerular ≥ 50 mL/min y que la ficha técnica de E/C/F/TDF indica que no debe iniciarse tratamiento en pacientes con cifras menores a 70 mL//min.
Figura 1. GS-US-292-0104 y GS-US-292-0111: Cambio medio de Crsrespecto al nivel basal

Efectos adversos de especial interés en poblaciones especiales
Pacientes con insuficiencia renal
En pacientes con insuficiencia renal, la prevalencia de proteinuria clínicamente significativa (relación proteína/creatinina >200 mg/g) y albuminuria (relación proteína/creatinina ≥ 30 mg/g)disminuyó de 42 % al inicio del tratamiento a 16 % en la semana 48, y de 49 % al inicio a 26 % en la semana 48, respectivamente. Esteefecto se pudo observar desde la primera semana del cambio aE/C/F/TAF desde un régimen que contuviera TDF.
También se notaron mejoras, globalmente, en la DMO en la semana 48 en pacientes que habían cambiado a E/C/F/TAF. Por grupo de tratamiento anterior, esta mejora fue mayor en los pacientes que no habían recibido regímenes con TDF previamente. En cambio, en la cohorte de pacientes naïve no se observó un aumento de la DMO en la semana 48.
Adolescentes
En el estudio de pacientes adolescentes naïve se incluyeron 50 pacientes, de los cuales todos recibieron E/C/F/TAF al menos durante 8 semanas, y 25 hasta la semana 48. El 36 % tuvo efectos adversos asociados al fármaco, la mayoría de grado 1 ó 2 de severidad, los más frecuentes fueron náuseas, dolor abdominal, vómitos y diarrea. Cuatro pacientes (8 %) tuvieron efectos adversos graves pero solo uno se asoció al fármaco (uveítis intermedia que se resolvió y no requirió interrumpir el tratamiento).
No se observaron cambios significativos en la DMO ni alteraciones en los parámetros hematológicos.
Se describieron ligeros aumentos de colesterol total, LDL yHDL en la semana 48.
La mediana de creatinina aumentó la primera semana, pero se mantuvo estable hasta la semana 24. Consecuentemente, el ClCr disminuyó la primera semana con un cambio en la mediana (Q1, Q3) de -13 (-26, 0) mL/min/1,73 m2disminuyendo a una mediana de cambio de – 15 (-30, 0) mL/min/1,73 m2en la semana 24. Se observaron proteinurias aisladas y transitorias.
DISCUSIÓN
El mecanismo de acción de E/C/F/TAF es el mismo que el de otros regímenes que incluyen estos principios activos. La diferencia más destacable la tiene el nuevo profármaco de tenofovir, tenofovir alafenamida y se encuentra a nivel farmacocinético. Estas variaciones farmacocinéticas tienen repercusiones positivas a nivel de seguridad.
La presentación en forma de combinación a dosis fijas en un único comprimido presenta las mismas ventajas que otras presentaciones disponibles similares. La simplificación del régimen por un lado, podría favorecer la adherencia en aquellos pacientes que vean difícil abordar una alta carga de comprimidos y frecuencia de administración y, por otro, podría mantener una carga viral suprimida.
Contiene principios activos de los grupos terapéuticos recomendados para el control del VIH, incluyendo dos ITIAN y un InInt. Existe un inconveniente del elvitegravir en comparación con los otros InInt, raltegravir y dolutegravir, que necesita ser potenciado para favorecer su actividad farmacológica. La potenciación con cobicistat implica la aparición de interacciones con otros fármacos, que pueden contraindicar el tratamiento concomitante si el paciente toma estos fármacos. Además, elvitegravir y dolutegravir no se recomiendan en el embarazo por ausencia de datos concluyentes, a diferencia del raltegravir que sí se puede utilizar en el embarazo.
La eficacia del E/C/F/TAF en pacientes adultos naïve se sustenta en ensayos clínicos que demuestran la no inferioridad respecto a su comparador principal, el E/C/F/TDF. La elección de este comparador se considera adecuada y permite además comparar los perfiles de seguridad del TDF y TAF.
En el ensayo de fase 2 y en uno de los ensayos pivotales, los análisis llevados a cabo por subgrupos indican que E/C/F/TAF tiene resultados algo inferiores a E/C/F/TDF en los subgrupos de pacientes con carga viral basal elevada y recuento basal de linfocitos CD4 bajo. Esta tendencia no se observó en el segundo ensayo pivotal y no identificó ninguna característica basal concreta que explicara este hallazgo. Estos resultados han quedado adecuadamente reflejados en la ficha técnica.
Igualmente, E/C/F/TAF ha demostrado el mantenimiento de la respuesta viral suprimida al cambiar de regímenes que contenían TDF más un tercer agente.
También se evaluó la eficacia en poblaciones especiales. Se demostró la eficacia en pacientes con insuficiencia renal instaurada leve o moderada, incluyendo pacientes con enfermedad renal crónica asociada o no al uso previo de TDF. El tamaño de la cohorte de pacientes con insuficiencia renal naïve fue demasiado pequeña como para obtener conclusiones relevantes.
Se ha demostrado la eficacia en una muestra pequeña de pacientes naïve adolescentes. Los datos sugieren respuestas al tratamiento similares a las observadas en adultos. Por otro lado, losincrementos observados en el recuento de CD4 son más variables que en los adultos, probablemente debido a que el tamaño de la muestra es inferior.
El perfil de seguridad del E/C/F/TAF es globalmente aceptable, con un porcentaje bajo de efectos adversos graves. Uno de los principales inconvenientes del uso de TDF son sus efectos adversos identificados a nivel óseo y renal, los cuales causan, en muchos casos, interrupciones y cambios en el tratamiento antirretroviral del paciente.
A nivel óseo, los datos de seguridad sugieren una mejoría en cuanto a la densidad mineral ósea frente a E/C/F/TDF. En los estudios sin comparador en insuficiencia renal y adolescentes, también se observa que DMO mejora o no se altera, respectivamente.
En cuanto a la seguridad renal, los datos sugieren también que TAF podría inducir menor daño renal observándose incluso una mejoría en los marcadores de daño renal (la proteinuria y albuminuria instauradas en pacientes con enfermedad renal crónica mejora a lo largo del tiempo, apreciándose el resultado a las pocas semanas de inicio).
Se debe tener en cuenta que los estudios no tienen potencia para evaluar eventos adversos tales como fallo renal o fractura. Además, se desconoce si estas mejoras se mantendrán en el tiempo, siendo necesario resultados de estudios a más largo plazo. Si bien es cierto que en los estudios 104 y 111, la combinación de E/C/F/TAF presenta diferencias estadísticamente significativas frente a E/C/F/TDF tanto a nivel de filtración glomerular (marcador de nefrotoxocidad) y de cambio en densidad mineral ósea, estas diferencias no sabemos si tendrán transcendencia clínica y si estas mejoras se mantendrán en el tiempo ya que se han estudiado a 96 semanas, siendo necesarios resultado de estudios a más largo plazo.
El incremento de lípidos es superior con E/C/F/TAF que con E/C/F/TDF, pero sin diferencias en los cambios del cociente Colesterol total/HDL. Estas diferencias sugieren que TAF carece del efecto hipolipemiante previamente descrito con TDF.
CONCLUSIÓN
La combinación de elvitegravir/cobicistat/ emtricitabina/tenofovir alafenamida está indicada para el tratamiento de adultos y adolescentes (de 12 años de edad o mayores con un peso corporal de al menos 35 kg) infectados con el virus VIH-1 sin ninguna mutación conocida asociada con resistencia a los inhibidores de la integrasa, emtricitabina o tenofovir.
Elvitegravir/cobicistat/ emtricitabina/tenofovir alafenamida constituye una alternativa terapéutica frente a elvitegravir/cobicistat/emtricitabina/tenofovir disoproxil. Los datos de seguridad sugieren también que TAF induce menor daño renal, lo que podría suponer una ventaja a largo plazo. Sin embargo, se debe tener en cuenta que sólo se disponen de datos a 96 semanas.
Además, es posible usarlo en pacientes adolescentes y pacientes con una insuficiencia renal leve o moderada (filtrado glomerular mayor o igual a 30 ml/min/1,73 m2).
CONSIDERACIONESFINALES DELGCPT
Dado que no se han encontrado diferencias clínicamente relevantes entre la eficacia y seguridad del medicamento evaluado y sus alternativas, la elección entre ellos se basará fundamentalmente en criterios de eficiencia.
REFERENCIAS
1. Global Report: UNAIDS report on the global AIDS epidemic2013. Noviembre de 2013.
2. All-cause mortality in treated HIV-infected adults with CD4≥500/mm3 compared with the general population: evidencefrom a large European observational cohort collaboration.
3. Documento de consenso de Gesida/Plan Nacional sobre el Sida respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana. Enero 2016. Disponible en:http://gesida-seimc.org/contenidos/guiasclinicas/2016/gesida-guiasclinicas-2016-tar.pdf.
4. Knobel H, Polo R, Escobar I (Coordinadores). Recomendaciones Gesida/SEFH/PNS para mejorar la adherencia al tratamiento antirretroviral (Actualización junio de 2008).
5. EPAR de Genvoya®. Disponible en:http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004042/WC500197863.pdf. (Consultado 11/01/2016).
6. Ficha técnica de Genvoya®. Disponible en:http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/004042/WC500197861.pdf. (Consultado el 11/01/2016).
7. Sax PE, Whol D, Yin MT, Post F, DeJesus E, Saag M et al.Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, forinitial treatment of HIV-1 infection:two randomised, double-blind, Phase 3, non-inferiority trials. Lancet. 2015385(9987):2,606-2,615.
8. Pozniak A, Arribas JR, Gathe J, Gupta SK, Post FA, Bloch Met al; GS-US-292-0112 Study Team. Switching to Tenofovir Alafenamide, Coformulated With Elvitegravir, Cobicistat, and Emtricitabine, in HIV-Infected Patients With Renal Impairment:48-Week Results From a Single-Arm, Multicenter, Open-LabelPhase 3 Study. J Acquir Immune Defic Syndr. 2016 Apr15;71(5):530-7. doi: 10.1097/QAI.0000000000000908.
9. Mills A, Arribas JR, Andrade-Villanueva J, DiPerri G, Van Lunzen J et al, GS-US-292-0109 team.. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in antiretroviral regimens for virologically suppressed adults with HIV-1 infection: a randomised, active-controlled, multicentre, open-label, phase 3, non-inferiority study. Lancet Infect Dis.2016 Jan;16(1):43-52. doi: 10.1016/S1473-3099(15)00348-5. Epub 2015 Nov 2.
10. Agarwal K, et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol. 2015 Mar;62(3):533-40.
11. Buti M et al. A Phase 3 Study of TAF Compared with TDF inPatients with HBeAg-negative, Chronic Hepatitis B: Week 48Efficacy and Safety Results. Presented at EASL 2016.
12. Gallant J, Brunetta J, Crofoot G, et al. Efficacy and Safety of Switching to Simpler Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Alafenamide (E/C/F/TAF) in HIV-1/Hepatitis B Coinfected Adults in North America and Japan (NCT02071082): Week 48 Results [Poster WELBPA13]. Paper presented at: 8th IAS Conference on HIV Pathogenesis, Treatment andPrevention; 19-22 July, 2015; Vancouver, British Columbia, Canada.
13. Wohl D., Oka S., Clumeck N., Clarke A, Brinson C, Stephens J. et al. A Randomized, Double-Blind Comparison of Tenofovir Alafenamide vs Tenofovir Disoproxil Fumarate, Each Coformulated With Elvitegravir, Cobicistat, and Emtricitabine, for Initial HIV-1 Treatment: Week 96 J Acquir Immune Defic Syndr 2016;72:58–64. doi: 10.1097/QAI.0000000000000940.
GRUPO DE EXPERTOS
(por orden alfabético)
Agencia Española de Medicamentos y Productos Sanitarios
Comunidad Autónoma del Principado de Asturias
Todos los expertos han realizado una declaración de conflictos de interés.
El Laboratorio Titular, la Sociedad Española de Farmacia Hospitalaria, la Sociedad Española de Farmacología Clínica, la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica, la Sociedad Española Interdisciplinaria del SIDA, Grupo de Trabajo sobre Tratamientos del VIH y Apoyo Positivo han tenido oportunidad de enviar comentarios al documento, si bien el texto final es el adoptado por el GCPT.
Fuente: Agencia Española de Medicamentos y Productos Sanitarios.
