ZEJULA 100 MG CAPSULAS DURAS

| ATC: Niraparib |
| PA: Niraparib tosilato monohidrato |
| EXC:
Lactosa monohidrato Tartrazina y otros. |
Envases
- Env. con 84
- DHSC: Medicamento de diagnóstico hospitalario sin cupón precinto
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica Medicamento Huérfano
Medicamento Huérfano- Fi: Medicamento incluido en la financiación del SNS
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 720555
- EAN13: 8470007205555
- Conservar en frío: No
- Env. con 56
- DHSC: Medicamento de diagnóstico hospitalario sin cupón precinto
- Dispensación sujeta a prescripción médica
- Medicamento Huérfano
- Fi: Medicamento incluido en la financiación del SNS
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 720556
- EAN13: 8470007205562
- Conservar en frío: No
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
ZEJULA 100 mg Cáps. dura
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene tosilato de niraparib monohidrato equivalente a 100 mg de niraparib.
Excipiente(s) con efecto conocido
Cada cápsula dura contiene 254,5 mg de lactosa monohidrato (ver sección 4.4).
La envoltura de cada cápsula dura también contiene 0,0172 mg de tartrazina (E 102) como agente colorante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. - FORMA FARMACÉUTICA
Cápsula dura (cápsula).
Cápsula dura de aproximadamente 22 mm × 8 mm; cuerpo blanco con "100 mg" impreso con tinta negra y cierre morado con "Niraparib" impreso con tinta blanca.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de ZEJULA 100 mg Cáps. dura
Zejula está indicado:
- como tratamiento de mantenimiento en monoterapia de pacientes adultas con cáncer de ovario epitelial avanzado (estadios FIGO III y IV) de alto grado, trompas de Falopio o peritoneal primario que están en respuesta (completa o parcial) tras completar una primera línea de quimioterapia basada en platino.
- como tratamiento de mantenimiento en monoterapia de pacientes adultas con cáncer de ovario epitelial seroso de alto grado, trompas de Falopio o peritoneal primario, en recaída, sensible a platino, que están en respuesta (completa o parcial) a la quimioterapia basada en platino.
4.2 - Posología y administración de ZEJULA 100 mg Cáps. dura
4.3 - Contraindicaciones de ZEJULA 100 mg Cáps. dura
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Lactancia materna (ver sección 4.6).
4.4 - Advertencias y Precauciones de ZEJULA 100 mg Cáps. dura
Reacciones adversas hematológicas
Se han notificado reacciones adversas hematológicas (trombocitopenia, anemia, neutropenia) en pacientes tratadas con Zejula (ver sección 4.8). Las pacientes con un menor peso corporal o un menor recuento de plaquetas basal pueden tener un mayor riesgo de sufrir trombocitopenia de Grado ≥ 3 (ver sección 4.2).
Para controlar los cambios clínicamente relevantes en cualquier parámetro hematológico durante el tratamiento se recomienda realizar hemogramas completos semanalmente durante el primer mes; seguidos de controles mensuales durante los siguientes 10 meses de tratamiento, y periódicamente después de este período (ver sección 4.2).
Si una paciente presenta toxicidad hematológica persistente grave incluyendo pancitopenia que no se resuelve en un plazo de 28 días después de la interrupción del tratamiento, se debe suspender la administración de Zejula.
A causa del riesgo de trombocitopenia, se deben usar con precaución los anticoagulantes y medicamentos con un efecto reductor conocido en el recuento de trombocitos (ver sección 4.8).
Síndrome mielodisplásico/leucemia mieloide aguda
Se han observado casos de síndrome mielodisplásico/leucemia mieloide aguda (SMD/LMA), incluyendo casos con desenlace fatal, en pacientes tratadas con Zejula en monoterapia o en combinación en ensayos clínicos y tras la comercialización (ver sección 4.8).
En ensayos clínicos, la duración del tratamiento con Zejula en las pacientes antes de presentar SMD/LMA varió de 0,5 meses a > 4,9 años. Los casos fueron característicos de SMD/LMA secundario, relacionado con el tratamiento antineoplásico. Todas las pacientes habían recibido líneas de quimioterapia basada en platino y muchas también habían recibido otros fármacos que dañan el ADN, así como radioterapia. Algunas pacientes tenían antecedentes de supresión de la médula ósea. En el ensayo NOVA, la incidencia de SMD/LMA fue mayor en la cohorte gBRCA mutada (7,4 %) que en la cohorte gBRCA no mutada (1,7 %).
En caso de sospecha de SMD/LMA o toxicidades hematológicas prolongadas, se debe derivar a la paciente a un hematólogo para una evaluación adicional. Si se confirma el diagnóstico de SMD/LMA, se debe suspender el tratamiento con Zejula y la paciente debe recibir el tratamiento apropiado.
Hipertensión, incluida la crisis hipertensiva
Se ha notificado hipertensión, incluida crisis hipertensiva, con el uso de Zejula (ver sección 4.8). Antes de empezar el tratamiento con Zejula se debe controlar adecuadamente la hipertensión preexistente. Durante el tratamiento con Zejula se debe controlar la presión arterial al menos semanalmente durante dos meses, después mensualmente durante el primer año y, después, de forma periódica. En las pacientes adecuadas se puede considerar el control de la presión arterial en el domicilio, informando a la paciente que debe contactar con su médico en caso de aumento de la presión arterial.
Se debe controlar la hipertensión con medicamentos antihipertensivos; si es necesario, también se debe ajustar la dosis de Zejula (ver sección 4.2). En el programa clínico, se realizaron mediciones de la presión arterial en el Día 1 de cada ciclo de 28 días, mientras la paciente permanecía en tratamiento con Zejula. En la mayoría de los casos, la hipertensión se controló adecuadamente con el tratamiento antihipertensivo habitual, con o sin el ajuste de la dosis de Zejula (ver sección 4.2). Se debe suspender el tratamiento con Zejula en caso de crisis hipertensiva o si la hipertensión clínicamente relevante no se puede controlar adecuadamente con tratamiento antihipertensivo.
Síndrome de encefalopatía posterior reversible (PRES)
Se han notificado casos de PRES en pacientes que reciben Zejula (ver sección 4.8). PRES es un trastorno neurológico raro y reversible que puede presentarse con síntomas que evolucionan rápidamente, como crisis convulsivas, cefalea, alteración del estado mental, deterioro visual o ceguera cortical, con o sin hipertensión asociada. El diagnóstico de PRES requiere confirmación mediante técnicas de imagen cerebral, preferiblemente por resonancia magnética (IRM).
En caso de PRES, se recomienda suspender el tratamiento con Zejula y tratar los síntomas específicos, incluida la hipertensión. Se desconoce la seguridad de reiniciar el tratamiento con Zejula en aquellas pacientes que han experimentado PRES previamente.
Embarazo/anticoncepción
No se debe usar Zejula durante el embarazo, ni en mujeres en edad fértil que no estén dispuestas a utilizar métodos anticonceptivos altamente efectivos durante el tratamiento y durante 6 meses después de recibir la última dosis de Zejula (ver sección 4.6). Se debe hacer una prueba de embarazo a todas las mujeres en edad fértil antes del tratamiento.
Insuficiencia hepática
Las pacientes con insuficiencia hepática grave podrían tener una mayor exposición a niraparib según los datos de pacientes con insuficiencia hepática moderada y deben ser cuidadosamente controladas (ver secciones 4.2 y 5.2).
Lactosa
Las cápsulas duras de Zejula contienen lactosa monohidrato. Los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
Tartrazina (E 102)
Este medicamento contiene tartrazina (E 102), que puede provocar reacciones alérgicas.
4.5 - Interacciones con otros medicamentos de ZEJULA 100 mg Cáps. dura
Interacciones farmacodinámicas
No se ha estudiado la combinación de niraparib con vacunas o agentes inmunosupresores.
Los datos sobre niraparib en combinación con medicamentos citotóxicos son limitados. Por lo tanto, se debe tener precaución si se usa niraparib en combinación con vacunas, agentes inmunosupresores o con otros medicamentos citotóxicos.
Interacciones farmacocinéticas
Efecto de otros medicamentos sobre niraparib
Niraparib como sustrato de los CYPs (CYP1A2 y CYP3A4)
Niraparib es un sustrato de las carboxilesterasas (CEs) y UDP-glucuronosiltransferasas (UGTs) in vivo. El metabolismo oxidativo del niraparib es mínimo in vivo. No se requiere un ajuste de la dosis de Zejula cuando se administra de forma concomitante con medicamentos con un efecto conocido de inhibición (p. ej., itraconazol, ritonavir y claritromicina) o inducción de enzimas del CYP (p. ej., rifampicina, carbamazepina y fenitoína).
Niraparib como sustrato de los transportadores de eflujo (P-gp, BCRP, BSEP, MRP2 y MATE1/2)
Niraparib es un sustrato de la glucoproteína P (P-gp) y de la Proteína de Resistencia del Cáncer de Mama (BCRP). Sin embargo, debido a su permeabilidad y biodisponibilidad elevadas, es poco probable el riesgo de interacciones clínicamente relevantes con medicamentos que inhiben estos transportadores. Por lo tanto, no se requiere un ajuste de la dosis de Zejula cuando se administra de forma concomitante con medicamentos con un efecto conocido de inhibición de la P-gp (p. ej., amiodarona, verapamilo) o de la BCRP (p. ej., osimertinib, velpatasvir y eltrombopag).
Niraparib no es un sustrato de la bomba de exportación de sales biliares (BSEP), o proteína 2 relacionada con resistencia a múltiples fármacos (MRP2). El principal metabolito primario, M1, no es un sustrato de la P-gp, BCRP, BSEP o MRP2. El niraparib no es un sustrato de extrusión de múltiples fármacos y toxinas (MATE) 1 o 2, mientras que M1 es un sustrato de ambos.
Niraparib como sustrato de transportadores de la captación hepática (OATP1B1, OATP1B3 y OCT1)
Ni niraparib ni el M1 son sustratos del polipéptido de transporte de aniones orgánicos 1B1 (OATP1B1), 1B3 (OATP1B3) o del transportador de cationes orgánicos 1 (OCT1). No se requiere un ajuste de la dosis de Zejula cuando se administra de forma concomitante con medicamentos con un efecto conocido de inhibición de transportadores de la captación del OATP1B1 o 1B3 (p. ej., gemfibrozilo, ritonavir) o del OCT1 (p. ej., dolutegravir).
El niraparib como sustrato de transportadores de la captación renal (OAT1, OAT3 y OCT2)
Ni el niraparib ni el M1 son sustratos del transportador de aniones orgánicos 1 (OAT1), 3 (OAT3) ni del transportador de cationes orgánicos 2 (OCT2). No se requiere un ajuste de la dosis de Zejula cuando se administra de forma concomitante con medicamentos con un efecto conocido de inhibición de transportadores de la captación del OATI (p. ej., probenecid), OAT3 (p. ej., probenecid, diclofenaco) u OCT2 (p. ej., cimetidina, quinidina).
Efecto de niraparib sobre otros medicamentos
Inhibición de los CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4)
Ni niraparib ni el M1 son inhibidores de ninguna de las enzimas CYP metabolizadoras de principios activos, como CYP1A1/2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4/5.
Aunque no se prevé la inhibición del CYP3A4 en el hígado, no se ha establecido el potencial de inhibición del CYP3A4 a nivel intestinal en la concentración correspondiente de niraparib. Por lo tanto, se recomienda precaución si niraparib se asocia a principios activos cuyo metabolismo es dependiente del CYP3A4 y, notablemente, los que tienen un margen terapéutico estrecho (p. ej., ciclosporina, tacrolimus, alfentanilo, ergotamina, pimozida, quetiapina y halofantrina).
Inhibición de la UDP-glucuronosiltransferasas (UGTs)
Niraparib no demostró efecto inhibidor frente las isoformas de UGT (UGT1A1, UGT1A4, UGT1A9 y UGT2B7) hasta 200 μM in vitro. Por lo tanto, el potencial de una inhibición clínicamente significativa de UGTs por niraparib es mínimo.
Inducción de los CYPs (CYP1A2 y CYP3A4)
Ni niraparib ni el M1 son inductores del CYP3A4 in vitro. In vitro, niraparib induce débilmente el CYP1A2 a concentraciones altas y no se podría descartar completamente la importancia clínica de este efecto. El M1 no es inductor del CYP1A2. Por lo tanto, se recomienda precaución si niraparib se asocia a principios activos cuyo metabolismo es dependiente del CYP1A2 y, notablemente, los que tienen un margen terapéutico estrecho (p. ej., clozapina, teofilina y ropinirol).
Inhibición de los transportadores de eflujo (P-gp, BCRP, BSEP, MRP2, y MATE1/2)
Niraparib no es inhibidor de la BSEP o MRP2. In vitro, niraparib inhibe la P-gp muy débilmente y la BCRP con una CI50 = 161 μM y 5,8 μM, respectivamente. Por lo tanto, aunque sea poco probable, no puede descartarse una interacción clínicamente importante, relacionada con una inhibición de estos transportadores de eflujo. Luego, se recomienda precaución si niraparib se asocia a sustratos de la BCRP (irinotecán, rosuvastatina, simvastatina, atorvastatina y metotrexato).
Niraparib es un inhibidor de MATE1 y de MATE2, con una CI50 de 0,18 μM y ≤ 0,14 μM, respectivamente. No se puede descartar el aumento de las concentraciones plasmáticas de medicamentos administrados de forma concomitante, que son sustratos de estos transportadores (p. ej., la metformina).
El principal metabolito primario, M1, no parece ser un inhibidor de la P-gp, BCRP, BSEP, MRP2 o MATE1/2.
Inhibición de los transportadores de la captación hepática (OATP1B1, OATP1B3 y OCT1)
Ni niraparib ni el M1 son inhibidores del polipéptido de transporte de aniones orgánicos 1B1 (OATP1B1) o 1B3 (OATP1B3).
In vitro, niraparib inhibe débilmente el transportador de cationes orgánicos 1 (OCT1) con una CI50 = 34,4 μM. Se recomienda precaución si niraparib se asocia a principios activos sometidos a transporte de captación por el OCT1, como, la metformina.
Inhibición de los transportadores de la captación renal (OAT1, OAT3 y OCT2)
Ni niraparib ni el M1 inhiben el transportador de aniones orgánicos 1 (OAT1), 3 (OAT3) y el transportador de cationes orgánicos 2 (OCT2).
Todos los estudios clínicos se han llevado a cabo solo en adultos.
4.6 - Embarazo y Lactancia de ZEJULA 100 mg Cáps. dura
Mujeres en edad fértil/anticoncepción en las mujeres
Las mujeres en edad fértil no deben quedarse embarazadas mientras reciben el tratamiento y no deben estar embarazadas al comienzo del tratamiento. Se debe hacer una prueba de embarazo a todas las mujeres en edad fértil antes del tratamiento. Las mujeres en edad fértil deben utilizar métodos anticonceptivos altamente efectivos durante el tratamiento y durante 6 meses después de recibir la última dosis de Zejula.
Embarazo
No hay datos o estos son limitados relativos al uso del niraparib en mujeres embarazadas. No se han realizado estudios de toxicidad para la reproducción y de toxicidad para el desarrollo en animales. Sin embargo, de acuerdo con su mecanismo de acción, niraparib podría causar daño embrionario o fetal, incluyendo muerte embrionaria y efectos teratogénicos, si se administra a una embarazada. No debe utilizarse Zejula durante el embarazo.
Lactancia
Se desconoce si niraparib o sus metabolitos se excretan en la leche materna. La lactancia materna está contraindicada durante la administración de Zejula y durante 1 mes después de recibir la última dosis (ver sección 4.3).
Fertilidad
No hay datos clínicos sobre la fertilidad. En ratas y perros se observó una disminución reversible de la espermatogénesis (ver sección 5.3).
4.7 - Efectos sobre la capacidad de conducción de ZEJULA 100 mg Cáps. dura
La influencia de Zejula sobre la capacidad para conducir y utilizar máquinas es moderada. Las pacientes que toman Zejula pueden presentar astenia, fatiga, mareos o dificultad para concentrarse. Las pacientes que presenten estos síntomas deben tener precaución al conducir o utilizar máquinas.
4.8 - Reacciones Adversas de ZEJULA 100 mg Cáps. dura
Resumen del perfil de seguridad
Las RAs de todos los grados que se produjeron en ≥ 10 % de las 851 pacientes que recibieron Zejula en monoterapia en los ensayos PRIMA (dosis de inicio de 200 mg o 300 mg) y NOVA fueron náuseas, anemia, trombocitopenia, fatiga, estreñimiento, vómitos, cefalea, insomnio, disminución del recuento de plaquetas, neutropenia, dolor abdominal, disminución del apetito, diarrea, disnea, hipertensión, astenia, mareos, disminución del recuento de neutrófilos, tos, artralgia, dolor de espalda, disminución del recuento de leucocitos y sofocos.
Las reacciones adversas graves más frecuentes > 1 % (frecuencia de aparición con el tratamiento) fueron trombocitopenia y anemia.
Tabla de reacciones adversas
Se han identificado las siguientes reacciones adversas en base a estudios clínicos y vigilancia poscomercialización en pacientes que recibieron Zejula en monoterapia (ver la Tabla 4). Las frecuencias de aparición de reacciones adversas se basan en datos de acontecimientos adversos obtenidos a partir de los estudios PRIMA y NOVA (dosis de inicio fija de 300 mg/día) donde la exposición de las pacientes es conocida y se definen como: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1 000 a < 1/100); raras (≥ 1/10 000 a < 1/1 000); y muy raras (< 1/10 000). Dentro de cada grupo de frecuencias, se presentan los efectos adversos en orden decreciente de gravedad.
Tabla 4. Tabla de reacciones adversas
| Clasificación por órganos y sistemas de MedDRA | Frecuencia de todos los grados de CTCAE* | Frecuencia de CTCAE* de grado 3 o 4 |
| Infecciones e infestaciones | Muy frecuentes Infección del tracto urinario Frecuentes Bronquitis, conjuntivitis | Poco frecuentes Infección del tracto urinario, bronquitis |
| Neoplasias benignas, malignas y no especificadas (incl quistes y pólipos) | Frecuentes Síndrome mielodisplásico/leucemia mieloide aguda** | Frecuentes Síndrome mielodisplásico/leucemia mieloide aguda** |
| Trastornos de la sangre y del sistema linfático | Muy frecuentes Trombocitopenia, anemia, neutropenia, leucopenia Poco frecuentes Pancitopenia, neutropenia febril | Muy frecuentes Trombocitopenia, anemia, neutropenia Frecuentes Leucopenia Poco frecuentes Pancitopenia, neutropenia febril |
| Trastornos del sistema inmunológico | Frecuentes Hipersensibilidad† | Poco frecuentes Hipersensibilidad |
| Trastornos del metabolismo y de la nutrición | Muy frecuentes Disminución del apetito Frecuentes Hipocalemia | Frecuentes Hipocalemia Poco frecuentes Disminución del apetito |
| Trastornos psiquiátricos | Muy frecuentes Insomnio Frecuentes Ansiedad, depresión, deterioro cognitivo†† Poco frecuentes Estado confusional | Poco frecuentes Insomnio, ansiedad, depresión, estado confusional |
| Trastornos del sistema nervioso | Muy frecuentes Cefalea, mareo Frecuentes Disgeusia Raras Síndrome de encefalopatía posterior reversible (PRES)** | Poco frecuentes Cefalea |
| Trastornos cardiacos | Muy frecuentes Palpitaciones Frecuentes Taquicardia | |
| Trastornos vasculares | Muy frecuentes Hipertensión Raras Crisis hipertensivas | Frecuentes Hipertensión |
| Trastornos respiratorios, torácicos y mediastínicos | Muy frecuentes Disnea, tos, nasofaringitis Frecuentes Epistaxis Poco frecuentes Neumonitis | Poco frecuentes Disnea, epistaxis, neumonitis |
| Trastornos gastrointestinales | Muy frecuentes Náuseas, estreñimiento, vómitos, dolor abdominal, diarrea, dispepsia Frecuentes Boca seca, distensión abdominal, inflamación de las mucosas, estomatitis | Frecuentes Náuseas, vómitos, dolor abdominal Poco frecuentes Diarrea, estreñimiento, inflamación de las mucosas, estomatitis, boca seca |
| Trastornos de la piel y del tejido subcutáneo | Frecuentes Fotosensibilidad, erupción | Poco frecuentes Fotosensibilidad, erupción |
| Trastornos musculoesqueléticos y del tejido conjuntivo | Muy frecuentes Dolor de espalda, artralgia Frecuentes Mialgia | Poco frecuentes Dolor de espalda, artralgia, mialgia |
| Trastornos generales y alteraciones en el lugar de administración | Muy frecuentes Fatiga, astenia Frecuentes Edema periférico | Frecuentes Fatiga, astenia |
| Exploraciones complementarias | Frecuentes Elevación de la gamma-glutamiltransferasa, AST elevada, aumento de creatinina en sangre, ALT elevada, aumento de la fosfatasa alcalina en sangre, disminución de peso | Frecuentes Elevación de la gamma-glutamiltransferasa, ALT elevada Poco frecuentes AST elevada, aumento de la fosfatasa alcalina en sangre |
* CTCAE=Criterios Comunes de Terminología para Eventos Adversos versión 4.02
** Basado en datos de los ensayos clínicos de niraparib. No se limita solamente al estudio pivotal en monoterapia ENGOT-OV16.
† Incluye hipersensibilidad, hipersensibilidad al fármaco, reacción anafiláctica, erupción farmacológica, angioedema y urticaria.
†† Incluye deterioro de la memoria, deterioro de la concentración.
Las reacciones adversas observadas en el grupo de pacientes a las que se les administró una dosis de inicio de 200 mg de Zejula según el peso o el recuento de plaquetas basal fueron de frecuencia similar o menor en comparación con el grupo al que se les administró una dosis de inicio fija de 300 mg (Tabla 4).
Consulte a continuación para obtener información específica sobre la frecuencia de trombocitopenia, anemia y neutropenia.
Descripción de reacciones adversas seleccionadas
Las reacciones adversas hematológicas (trombocitopenia, anemia, neutropenia), incluidos diagnósticos clínicos y/o valores analíticos, se produjeron generalmente en un momento temprano durante el tratamiento con niraparib y la incidencia disminuyó con el tiempo.
En los estudios NOVA y PRIMA, las pacientes elegibles para el tratamiento con Zejula tenían los siguientes parámetros hematológicos antes del inicio del tratamiento: recuento absoluto de neutrófilos (RAN) ≥ 1.500 células/μl; plaquetas ≥ 100.000 células/μl y hemoglobina ≥ 9 g/dl (NOVA) o ≥ 10 g/dl (PRIMA). En el programa clínico, las reacciones adversas hematológicas se manejaron con seguimiento analítico y ajuste de dosis (ver sección 4.2).
En PRIMA, a las pacientes a las que se les administró una dosis de inicio de Zejula basada en el peso o el recuento de plaquetas basal, se les redujeron la trombocitopenia, anemia y neutropenia Grado ≥ 3 del 48 % al 21 %, del 36 % al 23 % y del 24 % al 15 % respectivamente, en comparación con el grupo al que se administró una dosis de inicio fija de 300 mg. La suspensión debida a trombocitopenia, anemia y neutropenia ocurrió, en el 3 %, 3 % y 2 % de las pacientes respectivamente.
Trombocitopenia
En PRIMA, el 39 % de las pacientes tratadas con Zejula experimentaron trombocitopenia de Grado 3/4 en comparación con el 0,4 % de las pacientes que recibieron placebo con una mediana de tiempo desde la primera dosis hasta el primer acontecimiento de 22 días (rango: 15 a 335 días) y con una mediana de duración de 6 días (rango: 1 a 374 días). Se produjo la suspensión del tratamiento debido a trombocitopenia en el 4 % de las pacientes que recibieron niraparib.
En NOVA, aproximadamente el 60 % de las pacientes que recibieron Zejula presentaron trombocitopenia de cualquier grado y el 34 % de las pacientes presentaron trombocitopenia de Grado 3/4. En pacientes con un recuento plaquetario inferior a 180 × 109/l, se produjo trombocitopenia de cualquier grado y de Grado 3/4 en el 76 % y en el 45 % de las pacientes, respectivamente. Las medianas del tiempo hasta la aparición de trombocitopenia, con independencia del grado, y de trombocitopenia de Grado 3/4 fueron de 22 y 23 días, respectivamente. La tasa de incidencias nuevas de trombocitopenia después de hacer modificaciones intensivas de la dosis durante los dos primeros meses de tratamiento a partir del Ciclo 4 fue de 1,2 %. La mediana de la duración de los episodios de trombocitopenia de cualquier grado fue de 23 días y la mediana de duración de la trombocitopenia de Grado 3/4 fue de 10 días. Las pacientes tratadas con Zejula que presentan trombocitopenia podrían tener un aumento del riesgo de hemorragia. En el programa clínico, se abordó la trombocitopenia con controles analíticos, modificación de la dosis y transfusión de plaquetas en caso de ser necesario (ver sección 4.2). Se produjo la suspensión del tratamiento a causa de acontecimientos de trombocitopenia (trombocitopenia y disminución del recuento plaquetario) en aproximadamente el 3 % de las pacientes.
En el estudio NOVA, 48 de 367 (13 %) pacientes experimentaron hemorragia con trombocitopenia concurrente; todos los eventos hemorrágicos concurrentes con trombocitopenia fueron de gravedad Grado 1 o 2, excepto un evento de petequias y hematoma de Grado 3 observado simultáneamente con una reacción adversa grave de pancitopenia. La trombocitopenia ocurrió con mayor frecuencia en pacientes cuyo recuento de plaquetas basal era inferior a 180 × 109/l. Aproximadamente el 76 % de las pacientes que recibieron Zejula con plaquetas basales más bajas (<180 × 109/l) sufrieron trombocitopenia de cualquier grado, y el 45 % de las pacientes tuvieron trombocitopenia de Grado 3/4. Se ha observado pancitopenia en <1 % de las pacientes que recibieron niraparib.
Anemia
En PRIMA, el 31 % de las pacientes tratadas con Zejula tuvieron anemia de Grado 3/4 en comparación con el 2 % de las pacientes que recibieron placebo con una mediana de tiempo desde la primera dosis hasta el primer acontecimiento de 80 días (rango: de 15 a 533 días) y con una mediana de duración de 7 días (rango: de 1 a 119 días). Se produjo la suspensión del tratamiento debido a anemia en el 2 % de las pacientes que recibieron niraparib.
En NOVA, aproximadamente el 50 % de las pacientes presentaron anemia de algún grado y el 25 % presentaron anemia de Grado 3/4. La mediana del tiempo hasta el comienzo de la anemia de cualquier grado fue de 42 días, y de 85 días en el caso de los acontecimientos de Grado 3/4. La mediana de la duración de la anemia de cualquier grado fue de 63 días, y de 8 días, en el caso de los acontecimientos de Grado 3/4. La anemia de cualquier grado podría persistir durante el tratamiento con Zejula. En el programa clínico, la anemia se trató con controles analíticos, ajuste de la dosis (ver sección 4.2), y cuando se requirió, con transfusiones de hematíes. Se produjo la suspensión del tratamiento a causa de la anemia en el 1 % de las pacientes.
Neutropenia
En PRIMA, el 21 % de las pacientes tratadas con Zejula tuvieron neutropenia de Grado 3/4 en comparación con el 1 % de las pacientes que recibieron placebo con una mediana de tiempo desde la primera dosis hasta el primer acontecimiento de 29 días (rango: de 15 a 421 días) y con una mediana de duración de 8 días (rango: de 1 a 42 días). Se produjo la suspensión del tratamiento debido a neutropenia en el 2 % de las pacientes que recibieron niraparib.
En NOVA, aproximadamente el 30 % de las pacientes que recibieron Zejula presentaron neutropenia de cualquier grado y el 20 % de las pacientes presentaron neutropenia de Grado 3/4. La mediana del tiempo hasta el comienzo de la neutropenia de cualquier grado fue de 27 días, y de 29 días, en el caso de los acontecimientos de Grado 3/4. La mediana de la duración de la neutropenia de cualquier grado fue de 26 días, y de 13 días, en el caso de los acontecimientos de Grado 3/4. Además, se administró Factor Estimulante de Colonias de Granulocitos (G-CSF) a aproximadamente el 6 % de las pacientes tratadas con niraparib como tratamiento concomitante de la neutropenia. Se produjo la suspensión del tratamiento a causa de neutropenia en el 2 % de las pacientes.
Síndrome mielodisplásico/leucemia mieloide aguda
En estudios clínicos, se produjo SMD/LMA en el 1 % de las pacientes tratadas con Zejula, donde el 41 % de casos tuvo un desenlace fatal. La incidencia fue mayor en pacientes con cáncer de ovario en recaída, que habían recibido 2 o más líneas de tratamiento previo de quimioterapia basada en platino y con gBRCAmut tras 75 meses de seguimiento de supervivencia. Todas las pacientes tenían factores que contribuían al potencial desarrollo de SMD/LMA, habiendo recibido quimioterapia previa basada en platino. Muchas también habían recibido otros agentes dañinos del ADN y radioterapia. La mayoría de las notificaciones fueron en portadoras de gBRCAmut. Algunas de las pacientes tenían historial de cáncer previo o de supresión de la médula ósea.
En el estudio PRIMA, la incidencia de SMD/LMA fue de 0,8 % en pacientes que recibieron Zejula y 0,4 % en pacientes que recibieron placebo.
En el estudio NOVA en pacientes con cáncer de ovario en recaída, que habían recibido al menos 2 líneas previas de quimioterapia basada en platino, la incidencia global de SMD/LMA fue de 3,8 % en las pacientes que recibieron Zejula y de 1,7 % en las pacientes que recibieron placebo con un seguimiento de 75 meses. En las cohortes gBRCAmut y gBRCA no mutada, la incidencia de SMD/LMA fue de 7,4 % y de 1,7 % en pacientes que recibieron Zejula y 3,1 % y 0,9 % en pacientes que recibieron placebo, respectivamente.
Hipertensión
En PRIMA, se produjo hipertensión de Grado 3/4 en el 6 % de las pacientes tratadas con Zejula en comparación con el 1 % de las pacientes que recibieron placebo con una mediana de tiempo desde la primera dosis hasta el primer acontecimiento de 50 días (rango: de 1 a 589 días) y con una mediana de duración de 12 días (rango: de 1 a 61 días). No se suspendió el tratamiento debido a hipertensión en ningún paciente.
En NOVA, en el 19,3 % de las pacientes tratadas con Zejula se produjo hipertensión de cualquier grado. En el 8,2 % de las pacientes se produjo hipertensión de Grado 3/4. La hipertensión se trató fácilmente con medicamentos antihipertensivos. Se produjo la suspensión del tratamiento a causa de la hipertensión en < 1 % de las pacientes.
Población pediátrica
No se ha llevado a cabo ningún estudio en pacientes pediátricas.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 - Sobredosificación de ZEJULA 100 mg Cáps. dura
No existe ningún tratamiento específico en caso de sobredosis de Zejula y no se han establecido los síntomas de sobredosis. En caso de sobredosis, los facultativos deben seguir las medidas generales de apoyo y deben administrar tratamiento sintomático.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de ZEJULA 100 mg Cáps. dura
Grupo farmacoterapéutico: agentes antineoplásicos, otros agentes antineoplásicos, código ATC: L01XK02.
Mecanismo de acción y efectos farmacodinámicos
Niraparib es un inhibidor de las enzimas de la poli (ADP-ribosa) polimerasa (PARP), PARP-1 y PARP-2, que cumplen una función en la reparación del ADN. Los estudios in vitro han demostrado que la citotoxicidad inducida por niraparib puede suponer la inhibición de la actividad enzimática de la PARP y un aumento de la formación de complejos de PARP-ADN que tienen como resultado el daño del ADN, apoptosis y muerte celular. Se observó un aumento de la citotoxicidad inducida por niraparib en líneas de células tumorales con o sin deficiencias en los genes supresores de los tumores de cáncer de mama (BRCA) 1 y 2. En tumores de xenoinjerto derivados de pacientes (PDX) con cáncer ortotópico, de ovario epitelial seroso de alto grado, que crecieron en ratones, se ha demostrado que niraparib disminuye el crecimiento tumoral en las mutaciones BRCA 1 y 2, en BRCA no mutadas pero deficiente en recombinación homóloga (HR) y en tumores que son BRCA no mutadas y sin deficiencia detectable de HR.
Eficacia clínica y seguridad
Tratamiento de mantenimiento de primera línea para el cáncer de ovario
PRIMA es un estudio fase III, doble ciego, controlado con placebo en el que las pacientes (n = 733) en respuesta completa o parcial a una primera línea de quimioterapia basada en platino fueron aleatorizadas 2:1 a niraparib o placebo. PRIMA empezó con una dosis de inicio de 300 mg una vez al día en 475 pacientes (de las cuales 317 fueron aleatorizadas al grupo de niraparib vs 158 al grupo de placebo) en ciclos continuos de 28 días. La dosis de inicio en PRIMA se modificó en la Enmienda 2 del Protocolo. A partir de ese momento, a las pacientes que inicialmente tenían un peso corporal ≥ 77 kg y un recuento de plaquetas basal ≥ 150 000/μl se les administró diariamente 300 mg de niraparib (n = 34) o placebo (n = 21), mientras que a las pacientes que inicialmente tenían un peso corporal <77 kg o un recuento de plaquetas basal < 150 000/μl se les administró diariamente 200 mg de niraparib (n = 122) o placebo (n = 61).
Las pacientes fueron aleatorizadas después de completar una primera línea de quimioterapia basada en platino con/sin cirugía. Las pacientes fueron aleatorizadas en las 12 semanas siguientes al primer día del último ciclo de quimioterapia. Las pacientes recibieron ≥6 y ≤9 ciclos de terapia basada en platino. Después de la cirugía de intervalo, las pacientes recibieron ≥2 ciclos post operatorios de terapia basada en platino. Las pacientes que habían recibido bevacizumab con quimioterapia pero que no pudieron recibir bevacizumab como terapia de mantenimiento no fueron excluidas del estudio. Las pacientes no podían haber recibido tratamiento previo con un inhibidor del PARP (iPARP), incluido niraparib. Las pacientes que recibieron quimioterapia neoadyuvante seguida de cirugía de intervalo podían tener enfermedad residual visible o no tener enfermedad residual. Se excluyeron las pacientes con enfermedad en estadio III que tenían citorreducción completa (es decir, sin enfermedad residual visible) después de la cirugía citorreductora primaria. La aleatorización se estratificó según la mejor respuesta durante el régimen de platino de primera línea (respuesta completa versus respuesta parcial), quimioterapia neoadyuvante (QTNA) (Sí vs No); y el estado de deficiencia de recombinación homóloga (HRD) [positivo (HR deficiente) vs negativo (HR competente) o no determinado]. La prueba de HRD se realizó utilizando el test HRD en tejido tumoral obtenido en el momento del diagnóstico inicial. Los niveles de CA-125 debían estar en el rango normal (o una disminución de CA-125 > 90 %) durante el tratamiento de primera línea de la paciente y permanecer estables durante al menos 7 días.
Las pacientes comenzaron el tratamiento en el Ciclo 1/Día 1 (C1/D1) con 200 o 300 mg de niraparib o un placebo equivalente administrado una vez al día en ciclos continuos de 28 días. Las visitas clínicas se realizaron en cada ciclo (4 semanas ± 3 días).
La variable primaria fue la supervivencia libre de progresión (SLP), que se determinó mediante una revisión central independiente ciega (RCIC) según RECIST, versión 1.1. La supervivencia global (SG) fue un objetivo secundario clave. La SLP se realizó mediante un análisis jerarquizado: primero en la población con deficiencia de HR, luego en la población general. La mediana de edad de 62 años osciló entre 32 y 85 años entre las pacientes aleatorizadas con niraparib y de 33 a 88 años entre las pacientes aleatorizadas con placebo. El ochenta y nueve por ciento de todas las pacientes eran blancas. El sesenta y nueve por ciento de las pacientes aleatorizadas con niraparib y el 71 % de las pacientes aleatorizadas con placebo tenían un ECOG de 0 al inicio del estudio. En la población general, el 65 % de las pacientes tenía enfermedad en estadio III y el 35 % tenía enfermedad en estadio IV. En la población general, la localización del tumor primario en la mayoría de las pacientes (≥ 80 %) fue el ovario; la mayoría de las pacientes (> 90 %) tenían tumores con histología serosa. El sesenta y siete por ciento de las pacientes recibieron QTNA. El sesenta y nueve por ciento de las pacientes tuvo una respuesta completa a la primera línea de quimioterapia basada en platino. Un total de 6 pacientes con niraparib habían recibido bevacizumab como tratamiento previo para su cáncer de ovario.
PRIMA demostró una mejora estadísticamente significativa en la SLP de las pacientes aleatorizadas a niraparib en comparación con placebo en la población HR deficiente y en la población general (Tabla 5 y Figuras 1 y 2).
Los objetivos secundarios de eficacia incluyeron la SLP después de la primera terapia posterior (SLP2) y la SG (Tabla 5).
Tabla 5: Resultados de eficacia – PRIMA (determinado por RCIC)
| Población con deficiencia de HR | Población general | |||
| niraparib (N=247) | placebo (N=126) | niraparib (N=487) | placebo (N=246) | |
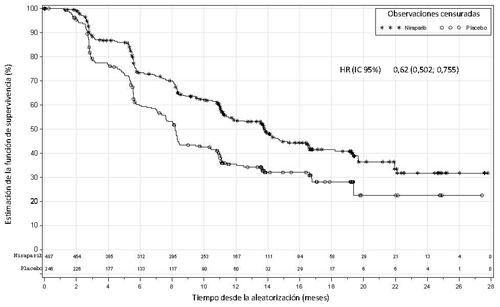
| Mediana SLP (IC 95 %) | 21,9 (19,3; NE) | 10,4 (8,1; 12,1) | 13,8 (11,5; 14,9) | 8,2 (7,3; 8,5) |
| Hazard ratio (IC 95 %) | 0,43 (0,31; 0,59) | 0,62 (0,50; 0,76) | ||
| Valor de p | <0,0001 | <0,0001 | ||
| SLP2 Hazard ratio (IC 95 %) | 0,84 (0,485; 1,453) | 0,81 (0,577; 1,139) | ||
| SG* Hazard ratio (IC 95 %) | 0,61 (0,265; 1,388) | 0,70 (0,44; 1,11) | ||
SLP = supervivencia libre de progresión; IC = intervalo de confianza; NE = no evaluable; SG = supervivencia global; SLP2 = SLP después de la primera terapia posterior.
* Supervivencia estimada a los dos años después de la aleatorización del 84 % para las pacientes que recibieron Zejula, en comparación con el 77 % para las pacientes que recibieron placebo en la población general, en el momento del análisis primario de la SLP.
Actualmente los datos de SLP2 y SG no son maduros.
Figura 1: Supervivencia libre de progresión en pacientes con tumores HR deficientes – PRIMA (población con intención de tratar, N=373)
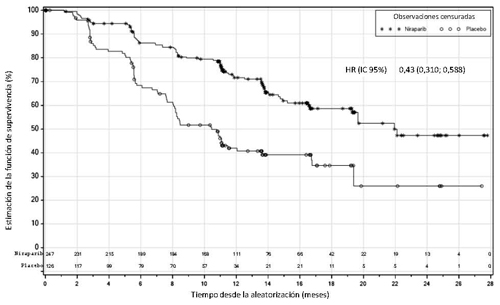
Figura 2: Supervivencia libre de progresión en la población general – PRIMA (población con intención de tratar, N=733)
Análisis por subgrupos
Dentro de la población HR deficiente, se observó un hazard ratio de 0,40 (IC del 95 %: 0,27; 0,62) en el subgrupo de pacientes con cáncer de ovario BRCAmut (N = 223). En el subgrupo de pacientes con HR deficiente sin una mutación en BRCA (N = 150), se observó un hazard ratio de 0,50 (IC del 95 %: 0,31; 0,83). En la población HR competente (N = 249), se observó un hazard ratio de 0,68 (IC del 95 %: 0,49; 0,94).
En un análisis exploratorio del subgrupo de pacientes a las que se les administró una dosis de 200 o 300 mg de Zejula según el peso o el recuento de plaquetas basales, se observó una eficacia comparable (SLP evaluada por el investigador) con un hazard ratio de 0,54 (IC del 95 %: 0,33; 0,91) en la población HR deficiente, y con un hazard ratio de 0,68 (IC del 95 %: 0,49; 0,94) en la población general. En el subgrupo de HR competente, la dosis de 200 mg pareció tener un efecto de tratamiento menor en comparación con la dosis de 300 mg.
Tratamiento de mantenimiento del cáncer de ovario en recaída sensible a platino
En un ensayo clínico internacional de fase III, aleatorizado, con doble ciego, comparado con placebo (NOVA) se estudió la seguridad y la eficacia de niraparib como tratamiento de mantenimiento en pacientes con cáncer de ovario epitelial seroso predominantemente de alto grado, de las trompas de Falopio o peritoneal primario, recidivado, que eran sensibles a platino, definida como la respuesta completa (RC) o la respuesta parcial (RP) durante más de seis meses desde su penúltimo tratamiento basado en platino. Para ser consideradas aptas para recibir tratamiento con niraparib, las pacientes debían estar en respuesta (RC o RP) después de finalizar la última quimioterapia basada en platino. Los valores de CA-125 debían ser normales (o una disminución > 90 % de CA-125 con respecto al valor basal) después de su último tratamiento con platino, y mantenerse estable durante al menos 7 días. Las pacientes no podían haber recibido un tratamiento previo con un iPARP, incluido Zejula. A las pacientes que cumplían los criterios se las asignó a una de las dos cohortes, de acuerdo con los resultados de una prueba de mutación del BRCA de la línea germinal (gBRCA). En cada cohorte, se asignó aleatoriamente a las pacientes con una asignación 2:1 de niraparib y placebo. Las pacientes fueron asignadas a la cohorte gBRCAmut en base a las muestras de sangre del análisis gBRCA, realizadas previamente a la aleatorización. La prueba de mutación BRCA tumoral (tBRCA) y HRD se realizó mediante el test HRD en tejido tumoral obtenido en el momento del diagnóstico inicial o de la recidiva.
La aleatorización dentro de cada cohorte se estratificó por el tiempo hasta la progresión después del penúltimo tratamiento con platino antes de la inclusión en el estudio (6 a < 12 meses y ≥ 12 meses); el uso o no de bevacizumab junto con la penúltima o la última pauta de platino, y la mejor respuesta durante la pauta más reciente de platino (respuesta completa y respuesta parcial).
Las pacientes empezaron el tratamiento en el Ciclo 1/Día 1 (C1/D1) con 300 mg de niraparib o con el correspondiente placebo, administrado una vez al día en ciclos continuados de 28 días. Se hicieron visitas clínicas en cada ciclo (4 semanas ± 3 días).
En el estudio NOVA, en el 48 % de las pacientes se interrumpió la dosis en el Ciclo 1.
Aproximadamente el 47 % de las pacientes volvió a empezar el tratamiento a una dosis reducida en el Ciclo 2.
La dosis usada con mayor frecuencia en las pacientes tratadas con niraparib en el estudio NOVA fue de 200 mg.
Se determinó la supervivencia libre de progresión (SLP) según RECIST (Criterios de Evaluación de la Respuesta en Tumores Sólidos, versión 1.1), o los signos y síntomas clínicos y el aumento de CA-125. La SLP se midió desde el momento de la aleatorización (que se produjo hasta 8 semanas después de la finalización de la pauta de quimioterapia) hasta la progresión de la enfermedad o la muerte.
El análisis de la eficacia principal en cuanto a la SLP se determinó mediante una revisión central independiente, ciega y se definió prospectivamente y se evaluó por separado en la cohorte con gBRCAmut y en la cohorte con gBRCA no mutada. Los análisis de supervivencia global (SG) fueron variables secundarias.
Las variables secundarias de eficacia incluyeron el intervalo libre de quimioterapia (ILQ), el tiempo hasta el primer tratamiento posterior (TPTP), la SLP después del primer tratamiento posterior (SLP2) y la SG.
En general, los datos demográficos, las características basales de la enfermedad y el historial de tratamiento previo estaban bien equilibrados entre los grupos de niraparib y placebo en el gBRCAmut (n = 203) y la de gBRCA no mutada (n = 350). Las medianas de edad variaron entre 57 y 63 años en todos los tratamientos y cohortes. La principal localización del tumor en la mayoría de las pacientes (> 80 %) en cada cohorte fue el ovario; la mayoría de las pacientes (> 84 %) tenía tumores con tipo histológico seroso. Una alta proporción de pacientes en ambos grupos de tratamiento y en ambas cohortes habían recibido tres o más líneas de quimioterapia anteriormente, incluidos el 49 % y el 34 % de las pacientes tratadas con niraparib en las cohortes con gBRCAmut y gBRCA no mutada, respectivamente. La mayoría de las pacientes tenían entre 18 y 64 años (78 %), eran caucásicos (86 %) y tenían un estado funcional ECOG de 0 (68 %).
En la cohorte con gBRCAmut, la mediana del número de ciclos de tratamiento fue mayor en el grupo que recibió niraparib que en el que recibió placebo (14 y 7 ciclos, respectivamente). Un mayor número de pacientes que recibieron niraparib continuaron el tratamiento durante más de 12 meses en comparación con el grupo de placebo (54,4 % y 16,9 % respectivamente). En la cohorte total con gBRCA no mutada, la mediana del número de ciclos de tratamiento fue más alta en el grupo que recibió niraparib que en el que recibió placebo (8 y 5 ciclos, respectivamente). Un mayor número de pacientes que recibieron niraparib continuaron el tratamiento durante más de 12 meses en comparación con el grupo de placebo (34,2 % y 21,1 %, respectivamente).
El estudio cumplió su objetivo principal de mejoría estadísticamente significativa de la SLP en cuanto al tratamiento de mantenimiento con niraparib en monoterapia, comparado con el placebo, en la cohorte con gBRCAmut, así como en la totalidad de la cohorte con gBRCA no mutada. La tabla 6 y Figuras 3 y 4 muestran los resultados correspondientes de la variable primaria de eficacia de SLP de las poblaciones (cohorte con gBRCAmut y la totalidad de la cohorte con gBRCA no mutada).
Tabla 6: Resumen de los resultados correspondientes al objetivo principal en el estudio NOVA.
| Cohorte con gBRCAmut | Cohorte con gBRCA no mutada | |||
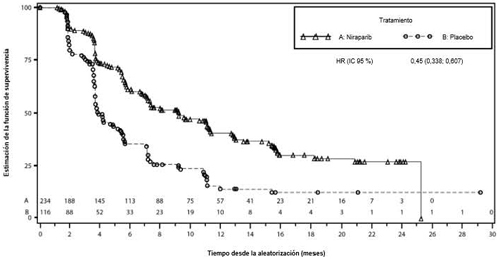
| niraparib (n = 138) | placebo (n = 65) | niraparib (n = 234) | placebo (n = 116) | |
| Mediana de la SLP (IC 95 %) | 21,0 (12,9; NE) | 5,5 (3,8; 7,2) | 9,3 (7,2; 11,2) | 3,9 (3,7; 5,5) |
| Valor de p | < 0,0001 | < 0,0001 | ||
| Hazard ratio (Nir:plac) (IC 95 %) | 0,27 (0,173; 0,410) | 0,45 (0,338; 0,607) | ||
SLP = supervivencia libre de progresión; IC = intervalo de confianza; NE = no evaluable.
Figura 3: Gráfico de Kaplan-Meier de la supervivencia libre de progresión en la cohorte de gBRCAmut evaluada mediante revisión central independiente (RCI) – NOVA (población con intención de tratar: n = 203)
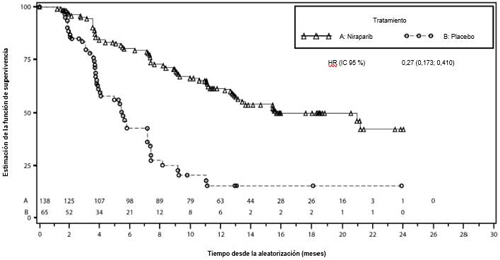
Figura 4: Gráfico de Kaplan-Meier de la supervivencia libre de progresión en la cohorte de gBRCA no mutada evaluada mediante revisión central independiente (RCI) – NOVA (población con intención de tratar, n = 350)
Variables secundarias de eficacia en NOVA
En el análisis final, la mediana de SLP2 en la cohorte gBRCAmut fue de 29,9 meses para las pacientes tratadas con niraparib en comparación con 22,7 meses para las pacientes que recibieron placebo (HR = 0,70; IC 95 %: 0,50; 0,97). La mediana de SLP2 en la cohorte gBRCA no mutada fue de 19,5 meses para las pacientes tratadas con niraparib en comparación con 16,1 meses para las pacientes que recibieron placebo (HR = 0,80; IC 95 %: 0,63; 1,02).
En el análisis final de supervivencia global, la mediana de SG en la cohorte gBRCAmut (n = 203) fue de 40,9 meses para las pacientes tratadas con niraparib en comparación con 38,1 meses para las pacientes que recibieron placebo (HR = 0,85; IC 95 %: 0,61; 1,20). La madurez de los datos en la cohorte gBRCAmut era del 76 %. La mediana de SG en la cohorte gBRCA no mutada (n = 350) fue de 31,0 meses para las pacientes tratadas con niraparib en comparación con 34,8 meses para las pacientes que recibieron placebo (HR = 1,06; IC 95 %: 0,81; 1,37). La madurez de los datos en la cohorte gBRCA no mutada era del 79 %.
Los resultados notificados por las pacientes (PRO) obtenidos de cuestionarios validados (FOSI y EQ5D) indican que las pacientes tratadas con niraparib no comunicaron ninguna diferencia respecto a las pacientes que recibieron placebo en las medidas asociadas con la calidad de vida (QoL).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Zejula en todos los grupos de la población pediátrica en el cáncer de ovario (excepto el rabdomiosarcoma y los tumores de células germinales).
5.2 - Propiedades farmacocinéticas de ZEJULA 100 mg Cáps. dura
Absorción
Después de la administración de una dosis única de 300 mg de niraparib en ayunas, niraparib fue medido en el plasma al cabo de 30 minutos, donde se alcanzó una concentración plasmática máxima media (Cmáx) de niraparib en aproximadamente 3 horas [804 ng/ml (% CV: 50,2 %)]. Después de varias dosis por vía oral de niraparib de 30 a 400 mg una vez al día, la acumulación del niraparib fue de aproximadamente 2 a 3 veces.
Las exposiciones sistémicas (Cmáx y AUC) al niraparib aumentaron en una manera proporcional a la dosis cuando la dosis de niraparib aumentó de 30 mg a 400 mg. La biodisponibilidad absoluta del niraparib es de aproximadamente el 73 %, lo que indica un efecto mínimo de primer paso. En un análisis farmacocinético poblacional de niraparib, se estimó la variabilidad interindividual de la biodisponibilidad con un coeficiente de variación (CV) del 31 %.
La administración concomitante de una comida rica en grasa no afectó de manera significativa a las propiedades farmacocinéticas de niraparib después de la administración de 300 mg de niraparib en cápsulas.
Se ha demostrado que las formulaciones en comprimido y cápsula son bioequivalentes. Tras la administración de un comprimido de 300 mg o de tres cápsulas de 100 mg de niraparib en 108 pacientes con tumores sólidos en condiciones de ayuno, los intervalos de confianza del 90 % de los cocientes de las medias geométricas de la Cmax, AUClast y AUC∞ para los comprimidos en comparación con las cápsulas estuvieron dentro de los límites de la bioequivalencia (0,80 y 1,25).
Distribución
Niraparib se unió de manera moderada a las proteínas en el plasma humano (83 %), principalmente con albúmina sérica. En un análisis farmacocinético poblacional de niraparib, el volumen de distribución aparente (Vd/F) fue de 1.311 L (basado en una paciente de 70 kg) en las pacientes con cáncer (CV 116 %), lo que indica una amplia distribución tisular de niraparib.
Biotransformación
Niraparib es metabolizado principalmente por carboxilesterasas (CE) para formar un metabolito inactivo principal, M1. En un estudio de balance de masas, M1 y M10 (los glucurónidos de M1 formados posteriormente) fueron los principales metabolitos circulantes.
Eliminación
Después de una dosis única de 300 mg niraparib por vía oral, el tiempo de semivida terminal media (t½) de niraparib osciló entre 48 y 51 horas (aproximadamente 2 días). En un análisis farmacocinético poblacional, el aclaramiento total aparente (CL/F) de niraparib fue de 16,5 l/h en las pacientes con cáncer (CV 23,4 %).
Niraparib se elimina principalmente por las vías hepatobiliar y renal. Después de una administración por vía oral de una dosis única de 300 mg de [14C] niraparib, se recuperó una media del 86,2 % (rango de 71 % a 91 %) de la dosis en la orina y las heces durante 21 días. La recuperación radioactiva en la orina fue del 47,5 % (rango de 33,4 % a 60,2 %), y en las heces, el 38,8 % (rango de 28,3 % a 47 %) de la dosis. En muestras agrupadas, recogidas durante 6 días, se recuperó el 40 % de la dosis en la orina, principalmente como metabolitos, y el 31,6 % de la dosis se recuperó en las heces, principalmente como niraparib inalterado.
Poblaciones especiales
Insuficiencia renal
En el análisis farmacocinético poblacional, las pacientes con insuficiencia renal leve (aclaramiento de creatinina 60-90 ml/min) y moderada (30-60 ml/min), tuvieron un aclaramiento de niraparib levemente reducido en comparación con individuos con una función renal normal (del 7-17 % mayor exposición en insuficiencia renal leve y del 17-38 % mayor exposición en insuficiencia renal moderada). No se considera que la diferencia en la exposición justifique un ajuste de dosis. En los estudios clínicos no se identificó a ninguna paciente con insuficiencia renal grave preexistente o con nefropatía terminal sometida a hemodiálisis (ver sección 4.2).
Insuficiencia hepática
En el análisis farmacocinético poblacional de los datos de los estudios clínicos en pacientes, la insuficiencia hepática leve preexistente (n = 155) no afectó al aclaramiento de niraparib. En un estudio clínico de pacientes con cáncer utilizando los criterios NCI-ODWG para clasificar el grado de insuficiencia hepática, el AUCinf de niraparib en pacientes con insuficiencia hepática moderada (n = 8) fue 1,56 (IC 90 %: 1,06; 2,30) veces el AUCinf de niraparib en pacientes con función hepática normal (n = 9) tras la administración de una dosis única de 300 mg. Se recomienda el ajuste de la dosis de niraparib en pacientes con insuficiencia hepática moderada (ver sección 4.2). La insuficiencia hepática moderada no afectó a la Cmáx de niraparib ni a la unión a proteínas de niraparib. No se han evaluado las propiedades farmacocinéticas de niraparib en las pacientes con insuficiencia hepática grave (ver secciones 4.2 y 4.4).
Peso, edad y raza
Los análisis farmacocinéticos poblacionales indicaron que el aumento de peso aumenta el volumen de distribución de niraparib. No se identificó impacto del peso en el aclaramiento y la exposición de niraparib. El peso corporal no justifica un ajuste de dosis, desde el punto de vista farmacocinético.
El análisis farmacocinético poblacional indicó que el aumento de la edad disminuía el aclaramiento de niraparib. La media de exposición en una paciente de 91 años se estimó un 23 % mayor que en una paciente de 30 años. No se considera que el impacto de la edad justifique un ajuste de dosis.
No hay datos suficientes entre razas para concluir su impacto sobre la farmacocinética de niraparib.
Población pediátrica
No se ha llevado a cabo ningún estudio para investigar las propiedades farmacocinéticas de niraparib en las pacientes pediátricas.
5.3 - Datos preclínicos sobre seguridad de ZEJULA 100 mg Cáps. dura
Farmacología de seguridad
In vitro, niraparib inhibió el transportador de la dopamina, DAT, a niveles de concentración inferiores a los niveles de exposición en seres humanos. En ratones, dosis únicas de niraparib aumentaron los niveles intracelulares de dopamina y de los metabolitos en el córtex. Se observó una disminución de la actividad psicomotriz en uno de dos estudios en ratones con una dosis única. Se desconoce la importancia clínica de estas observaciones. No se observó ningún efecto en los parámetros conductuales o neurológicos en estudios de toxicidad con dosis repetidas en ratas y perros, a concentraciones calculadas de exposición del SNC parecidas o inferiores a los valores esperados de exposición terapéutica.
Toxicidad con dosis repetidas
Se observó una disminución de la espermatogénesis en ratas y perros con niveles de exposición inferiores a los vistos en la clínica y fue en gran medida reversible en un plazo de 4 semanas después del cese de la administración.
Genotoxicidad
Niraparib no fue mutágeno en la prueba del ensayo de mutación bacteriana inversa (de Ames) pero fue clastógeno en un ensayo in vitro de aberración cromosómica en mamíferos y en un ensayo in vivo de micronúcleos en médula ósea de rata. Esta clastogenia es congruente con la inestabilidad genómica resultante de la farmacología principal de niraparib e indica genotoxicidad potencial en humanos.
Toxicología para la reproducción
No se han realizado estudios de toxicidad para la reproducción y para el desarrollo con niraparib.
Carcinogénesis
No se han realizado estudios de carcinogénesis con el niraparib.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de ZEJULA 100 mg Cáps. dura
Contenido de la cápsula
Estearato de magnesio
Lactosa monohidrato
Envoltura de la cápsula
Dióxido de titanio (E 171)
Gelatina
Azul brillante FCF (E 133)
Eritrosina (E 127)
Tartrazina (E 102)
Tinta para impresión
Goma laca (E 904)
Propilenglicol (E 1520)
Hidróxido de potasio (E 525)
Óxido de hierro negro (E 172)
Hidróxido de sodio (E 524)
Povidona (E 1201)
Dióxido de titanio (E 171)
6.2 - Incompatibilidades de ZEJULA 100 mg Cáps. dura
No procede.
6.3 - Período de validez de ZEJULA 100 mg Cáps. dura
3 años.
6.4 - Precauciones especiales de conservación de ZEJULA 100 mg Cáps. dura
No conservar a temperatura superior a 30 °C.
6.5 - Naturaleza y contenido del recipiente de ZEJULA 100 mg Cáps. dura
Blísteres perforados de dosis unitaria de Aclar/PVC/papel aluminio en cajas de 84 × 1, 56 × 1 y 28 × 1 cápsulas duras.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de ZEJULA 100 mg Cáps. dura
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
GlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublín 24
Irlanda
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/17/1235/001
EU/1/17/1235/002
EU/1/17/1235/003
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de noviembre de 2017
Fecha de la última renovación: 18 julio 2022
10. - FECHA DE LA REVISIÓN DEL TEXTO
05/2023
CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN
Medicamento sujeto a prescripción médica. Diagnóstico hospitalario sin cupón precinto. Reembolsable por el Sistema Nacional de Salud.
PRESENTACIONES Y PRECIO
Zejula 100mg cápsulas duras: envase de 56 cápsulas (C.N. 720556)
| PVL: 4.980 € | PVP: 5.035,91 € | PVP IVA: 5.237,35 € |
Zejula 100mg cápsulas duras: envase de 84 cápsulas (C.N. 720555)
| PVL: 7.470 € | PVP: 7.525,91 € | PVP IVA: 7.826,95 € |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
FT Promo ZEJULA CÁPSULAS - v15.0 (May 2023, FE GDSv5)
NP-ES-NRP-RPNT-230013 (v1) 08/2023

