 BRUKINSA 80 MG CAPSULAS DURAS
BRUKINSA 80 MG CAPSULAS DURAS
| ATC: Zanubrutinib |
| PA: Zanubrutinib |
Envases
- Env. con 120
- H: Medicamento de uso hospitalario
 Dispensación sujeta a prescripción médica
Dispensación sujeta a prescripción médica- Fi: Medicamento incluido en la financiación del SNS
- Facturable SNS: NO
- Comercializado: Si
- Situación: Alta
- Código Nacional: 732334
- EAN13: 8470007323341
- Conservar en frío: No
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
3. - FORMA FARMACÉUTICA
4. - DATOS CLÍNICOS
5. - PROPIEDADES FARMACOLÓGICAS
6. - DATOS FARMACÉUTICOS
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. - FECHA DE LA REVISIÓN DEL TEXTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
 1. - NOMBRE DEL MEDICAMENTO
1. - NOMBRE DEL MEDICAMENTO
BRUKINSA 80 mg Cáps. dura
2. - COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 80 mg de zanubrutinib
Para consultar la lista completa de excipientes, ver sección 6.1
3. - FORMA FARMACÉUTICA
Cápsula dura.
Cápsula dura opaca de color blanco o blanquecino de 22 mm de longitud, con “ZANU 80” marcado en tinta negra.
4. - DATOS CLÍNICOS
4.1 - Indicaciones Terapéuticas de BRUKINSA 80 mg Cáps. dura
BRUKINSA en monoterapia está indicado para el tratamiento de pacientes adultos con macroglobulinemia de Waldenström (MW) que han recibido al menos una terapia previa o como tratamiento de primera línea en pacientes que no son aptos para quimioinmunoterapia.
BRUKINSA en monoterapia está indicado para el tratamiento de pacientes adultos con linfoma de la zona marginal (LZM) que han recibido al menos una terapia previa con anticuerpos anti-CD20.
BRUKINSA en monoterapia está indicado para el tratamiento de pacientes adultos con leucemia linfocítica crónica (LLC).
4.2 - Posología y administración de BRUKINSA 80 mg Cáps. dura
4.3 - Contraindicaciones de BRUKINSA 80 mg Cáps. dura
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 - Advertencias y Precauciones de BRUKINSA 80 mg Cáps. dura
Hemorragia
Se han producido acontecimientos hemorrágicos graves y mortales en pacientes tratados con BRUKINSA en monoterapia. Se han notificado acontecimientos hemorrágicos de grado 3 y superior, que incluyen hemorragia intracraneal y gastrointestinal, hematuria y hemotórax, en los pacientes (ver sección 4.8). Se han producido acontecimientos hemorrágicos de cualquier grado, que incluyen púrpura y petequias, en pacientes con neoplasias malignas hematológicas. No se conoce bien el mecanismo de los acontecimientos hemorrágicos.
BRUKINSA puede aumentar el riesgo de hemorragia en los pacientes que reciben tratamientos antiagregantes plaquetarios o anticoagulantes y es necesario vigilar a los pacientes para detectar signos de hemorragia. Pueden precisarse modificaciones de la dosis en caso de reacciones adveras de grado 3 o superior según lo recomendado (ver sección 4.2). No se debe administrar de forma concomitante warfarina ni antagonistas de la vitamina K con BRUKINSA. Se debe controlar a los pacientes para la detección de signos y síntomas de hemorragia y vigilar los hemogramas completos. Se deben considerar los riesgos y los beneficios del tratamiento anticoagulante o antiagregante plaquetario cuando se administra conjuntamente con BRUKINSA. Se debe considerar el balance beneficio-riesgo de retirar el zanubrutinib durante 3 a 7 días antes y después de una intervención quirúrgica, en función del tipo de intervención y del riesgo de hemorragia.
Infecciones
Se han producido infecciones mortales y no mortales (infecciones bacterianas, víricas, micóticas o sepsis) e infecciones oportunistas (p. ej., infecciones por el virus del herpes, criptocócicas, por Aspergillus y por Pneumocystis jiroveci) en pacientes tratados con BRUKINSA en monoterapia. Se han producido infecciones de grado 3 o superior en los pacientes (ver sección 4.8). La infección de grado 3 o superior más frecuente fue la neumonía. También se han producido infecciones por reactivación del virus de la hepatitis B (VHB). Antes de iniciar el tratamiento con BRUKINSA se debe establecer el estado del VHB del paciente. Antes de iniciar tratamiento, se recomienda consultar a un médico experto en enfermedades hepáticas en el caso de pacientes con resultado positivo para el VHB o con serología positiva para la hepatitis B. Los pacientes deben controlarse y tratarse de acuerdo con los estándares médicos para prevenir la reactivación de la hepatitis B. Se debe considerar una profilaxis según la práctica habitual en los pacientes con un mayor riesgo de infecciones. Se debe vigilar a los pacientes para detectar signos y síntomas de infección y tratarlos adecuadamente.
Citopenias
Se han notificado citopenias de grado 3 o 4, incluidas neutropenia, trombocitopenia y anemia basadas en mediciones analíticas, en pacientes tratados con BRUKINSA en monoterapia (ver sección 4.8). Se deben vigilar los hemogramas completos mensualmente durante el tratamiento (ver sección 4.2).
Segundas neoplasias malignas primarias
Se han producido segundas neoplasias malignas primarias, que incluyen carcinoma no cutáneo, en pacientes con neoplasias malignas hematológicas tratados con BRUKINSA en monoterapia. La segunda neoplasia maligna primaria más frecuente fue el cáncer de piel (carcinoma de células basales y carcinoma de células escamosas de la piel). Se debe indicar a los pacientes que utilicen protección solar.
Fibrilación y aleteo auricular
Se ha producido fibrilación auricular y aleteo auricular en pacientes con neoplasias malignas hematológicas tratados con BRUKINSA en monoterapia, especialmente en pacientes con factores de riesgo cardíaco, hipertensión e infecciones agudas. Se deben controlar y tratar adecuadamente los signos y síntomas de fibrilación auricular y aleteo auricular.
Síndrome de lisis tumoral
El síndrome de lisis tumoral se ha notificado en casos infrecuentes con el tratamiento con zanubrutinib, especialmente en pacientes tratados por leucemia linfocítica crónica (LLC). Evaluar los riesgos pertinentes (p. ej., carga tumoral elevada o nivel de ácido úrico elevado) y tomar las medidas de precaución pertinentes. Controlar estrechamente a los pacientes y tratarlos adecuadamente.
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos muy efectivos durante el tratamiento con BRUKINSA (ver sección 4.6).
BRUKINSA contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente “exento de sodio”.
4.5 - Interacciones con otros medicamentos de BRUKINSA 80 mg Cáps. dura
El zanubrutinib se metaboliza principalmente mediante la enzima 3A del citocromo P450 (CYP3A).
Fármacos que pueden aumentar las concentraciones plasmáticas de zanubrutinib
El uso concomitante de BRUKINSA y medicamentos inhibidores potentes o moderados de CYP3A puede aumentar la exposición a zanubrutinib.
Inhibidores potentes de CYP3A
La administración concomitante de dosis múltiples de itraconazol (inhibidor potente de CYP3A) en voluntarios sanos aumentó la Cmáx de zanubrutinib en 2,6 veces y el AUC en 3,8 veces. La administración concomitante de dosis múltiples de los inhibidores potentes de CYP3A voriconazol y claritromicina en pacientes con neoplasias malignas de linfocitos B aumentó las exposiciones a zanubrutinib en 3,30 veces y en 1,92 veces para el AUC0-24h normalizada para la dosis y en 3,29 veces y 2,01 veces para la Cmáx normalizada para la dosis, respectivamente.
En caso de que se deba utilizar un inhibidor potente de CYP3A (p. ej., posaconazol, voriconazol, ketoconazol, itraconazol, claritromicina, indinavir, lopinavir, ritonavir, telaprevir), se debe reducir la dosis de BRUKINSA a 80 mg (una cápsula) durante el uso de inhibidor. Se debe vigilar al paciente de cerca para detectar toxicidad y seguir las directrices de modificación de la dosis según sea necesario (ver sección 4.2).
Inhibidores moderados de CYP3A
La administración concomitante de dosis múltiples de los inhibidores moderados de CYP3A fluconazol y diltiazem en pacientes con neoplasias malignas de linfocitos B aumentó las exposiciones a zanubrutinib en 1,88 veces y en 1,62 veces para el AUC0-24h normalizada para la dosis y en 1,81 veces y 1,62 veces para la Cmáx normalizada para la dosis, respectivamente.
En caso de que se deba utilizar un inhibidor moderado de CYP3A (p. ej., eritromicina, ciprofloxacino, diltiazem, dronedarona, fluconazol, verapamilo, aprepitant, imatinib, zumo de pomelo, naranjas amargas), se debe reducir la dosis de BRUKINSA a 160 mg (dos cápsulas) durante el uso del inhibidor. Se debe vigilar al paciente de cerca para detectar toxicidad y seguir las directrices de modificación de la dosis según sea necesario (ver sección 4.2).
Inhibidores leves de CYP3A
Las simulaciones en condiciones de ayuno sugirieron que los inhibidores leves de CYP3A (p. ej., ciclosporina y fluvoxamina) pueden aumentar el AUC de zanubrutinib en <1,5 veces. No se precisan ajustes de la dosis en combinación con inhibidores leves. Se debe vigilar al paciente de cerca para detectar toxicidad y seguir las directrices de modificación de la dosis según sea necesario.
El pomelo y las naranjas amargas se deben consumir con precaución durante el tratamiento con BRUKINSA, ya que contienen inhibidores moderados de CYP3A (ver sección 4.2).
Fármacos que pueden reducir las concentraciones plasmáticas de zanubrutinib
El uso concomitante de zanubrutinib e inductores potentes o moderados de CYP3A puede disminuir las concentraciones plasmáticas de zanubrutinib.
Inductores de CYP3A
La administración conjunta de dosis múltiples de rifampina (inductor potente de CYP3A) redujo la Cmáx de zanubrutinib en un 92 % y el AUC en un 93 % en sujetos sanos. Se debe evitar el uso concomitante con inductores potentes de CYP3A (p. ej., carbamazepina, fenitoína, rifampina, hierba de San Juan) e inductores moderados de CYP3A (p. ej., bosentán, efavirenz, etravirina, modafinilo, nafcilina) (ver sección 4.2). La administración conjunta de dosis múltiples de rifabutina (inductor moderado de CYP3A9 redujo la Cmáx de zanubrutinib en un 48 % y el AUC en un 44 % en sujetos sanos. Los inductores leves de CYP3A se pueden utilizar con precaución durante el tratamiento con BRUKINSA.
Fármacos reductores del ácido gástrico
No se observaron diferencias clínicamente significativas en la farmacocinética del zanubrutinib cuando se administra conjuntamente con fármacos reductores de ácido gástrico (inhibidores de la bomba de protones, antagonistas de los receptores de H2).
Fármacos cuyas concentraciones plasmáticas afecta el zanubrutinib
Zanubrutinib es un inductor leve de CYP3A y CYP2C19. El uso concomitante de zanubrutinib puede reducir las concentraciones plasmáticas de estos medicamentos que son sustratos.
Sustratos de CYP3A
La administración conjunta de dosis múltiples de zanubrutinib redujo la Cmáx de midalozam (sustrato de CYP3A) en el 30 % y el AUC en el 47 %. Los medicamentos con índice terapéutico estrecho metabolizados a través de CYP3A (p. ej., alfentanilo, ciclosporina, dihidroergotamina, ergotamina, fentanilo, pimozida, quinidina, sirólimus y tacrólimus) se deben utilizar con precaución, ya que el zanubrutinib puede reducir las exposiciones plasmáticas de estos medicamentos.
Sustratos de CYP2C19
La administración conjunta de dosis múltiples de zanubrutinib redujo la Cmáx de omeprazol (sustrato de CYP2C19) en el 20 % y AUC en el 36 %. Los medicamentos con índice terapéutico estrecho metabolizados a través de CYP2C19 (p. ej., S-mefentoína) se deben utilizar con precaución, ya que el zanubrutinib puede reducir las exposiciones plasmáticas de estos medicamentos.
Otros sustratos del CYP
No se observaron diferencias clínicamente significativas con la farmacocinética de la S-warfarina (sustrato de CYP2C9) cuando se administran conjuntamente con zanubrutinib.
Administración conjunta con sustratos/inhibidores transportadores
La administración conjunta de dosis múltiples de zanubrutinib aumentó la Cmáx de digoxina (sustrato de la gp-P) en el 34 % y el AUC en el 11 %. No hay diferencias clínicamente significativas en la farmacocinética de la rosuvastatina (sustrato de BCRP) cuando se administra conjuntamente con zanubrutinib.
La administración conjunta de sustratos orales de P-gp con un índice terapéutico estrecho (p. ej., digoxina) se debe realizar con precaución, ya que el zanubrutinib puede aumentar sus concentraciones.
4.6 - Embarazo y Lactancia de BRUKINSA 80 mg Cáps. dura
Mujeres en edad fértil/anticoncepción en mujeres
Sobre la base de los hallazgos en animales, BRUKINSA puede provocar daño fetal cuando se administra a embarazadas (ver sección 5.3). Las mujeres deben evitar quedarse embarazadas durante el tratamiento con BRUKINSA y hasta 1 mes después del fin del tratamiento. Por tanto, las mujeres en edad fértil deben utilizar un método anticonceptivo muy eficaz mientras estén tomando BRUKINSA y hasta 1 mes después de interrumpir el tratamiento. Actualmente se desconoce si zanubrutinib reduce la eficacia de los anticonceptivos hormonales; por tanto, las mujeres que los utilicen deben añadir un método de barrera. Se recomienda realizar pruebas de embarazo a las mujeres en edad fértil antes de iniciar el tratamiento.
Embarazo
No debe utilizarse BRUKINSA durante el embarazo. No hay datos relativos al uso de BRUKINSA en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
Lactancia
Se desconoce si el zanubrutinib o sus metabolitos se excretan en la leche materna y no se llevaron a cabo estudios clínicos. No pueden excluirse los riesgos para los bebés alimentados con leche materna. Durante el tratamiento con BRUKINSA debe suspenderse la lactancia.
Fertilidad
No se detectaron efectos en la fertilidad masculina o femenina en ratas, pero se observaron anomalías morfológicas en el esperma y aumento de las pérdidas posteriores a la implantación con 300 mg/kg/día (ver sección 5.3).
4.7 - Efectos sobre la capacidad de conducción de BRUKINSA 80 mg Cáps. dura
La influencia de BRUKINSA sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Se ha notificado fatiga, mareos y astenia en algunos pacientes que tomaban BRUKINSA y esto debe tenerse en cuenta al evaluar la capacidad del paciente para conducir u operar máquinas.
4.8 - Reacciones Adversas de BRUKINSA 80 mg Cáps. dura
Resumen del perfil de seguridad
Las reacciones adversas más frecuentes (≥20 %) fueron infección de las vías respiratorias altas§ (33 %), moretones§ (30 %), neutropenia§ (28 %), hemorragia/hematoma§ (27 %), erupción cutánea§ (23 %) y dolor musculoesquelético§ (23 %) (tabla 3).
Las reacciones adversas de grado 3 o superior más frecuentes (>5 %) fueron neutropenia§ (19 %), neumonía§ (9 %), hipertensión (7 %) y trombocitopenia§ (6 %).
De los 1150 pacientes tratados con zanubrutinib, el 2,9 % de los pacientes suspendió el tratamiento debido a reacciones adversas. La reacción adversa más frecuente que provocó la suspensión del tratamiento fue neumonía§ (1,4 %). En el 5,7 % de los pacientes se produjeron reacciones adversas que provocaron la reducción de la dosis.
Tabla de reacciones adversas
El perfil de seguridad se basa en los datos agrupados de 1550 pacientes con neoplasias malignas de células B, incluyendo pacientes con leucemia linfocítica crónica (N = 938), macroglobulinemia de Waldenström (N = 249), linfoma de células del manto (N = 140), linfoma de la zona marginal (N = 93), linfoma folicular (N = 59) y otros tipos de neoplasias malignas de células B (N = 71) tratados con BRUKINSA en estudios clínicos con una mediana de la duración de la exposición de 22,95 meses.
A continuación se presentan las reacciones adversas de los pacientes tratados con BRUKINSA para las neoplasias malignas de células B por clasificación de órganos del sistema y grupo de frecuencia. Las frecuencias se definen como: muy frecuentes (≥1/10), frecuentes (de ≥1/100 a <1/10), poco frecuentes (de ≥1/1.000 a <1/100), raras (de ≥1/10 000 a <1/1000), muy raras (<1/10 000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada categoría de frecuencia.
Tabla 3: Las reacciones adversas notificadas en los estudios clínicos en pacientes con neoplasias malignas de células B
| SOC del MedDRA | Términos del MedDRA | Todos los grados*(%) | Grado 3 o superior (%) |
| Infecciones e infestaciones | Infección de las vías respiratorias altas§ | Muy frecuentes (33) | 2 |
| Neumonía§# | Muy frecuentes (18) | 9 | |
| Neumonía | Muy frecuentes (12) | 7 | |
| Infección de las vías respiratorias bajas | Frecuentes (5) | <1 | |
| Infección urinaria | Muy frecuentes (12) | 2 | |
| Bronquitis | Frecuentes (4) | <1 | |
| Reactivación de la hepatitis B | Raras (<1) | <1 | |
| Trastornos de la sangre y del sistema linfático | Neutropenia§ | Muy frecuentes (28) | 19 |
| Neutropenia febril | Frecuentes (1) | 1 | |
| Trombocitopenia§ | Muy frecuentes (16) | 6 | |
| Anemia§ | Muy frecuentes (14) | 5 | |
| Trastornos del sistema nervioso | Mareo§ | Muy frecuentes (11) | <1 |
| Trastornos cardiacos | Fibrilación y aleteo auriculares | Frecuentes (3) | 1 |
| Trastornos vasculares | Moretones§ | Muy frecuentes (30) | <1 |
| Contusión | Muy frecuentes (18) | 0 | |
| Petequias | Frecuentes (7) | <1 | |
| Púrpura | Frecuentes (5) | <1 | |
| Equimosis | Frecuentes (2) | <1 | |
| Hemorragia/Hematoma§ # | Muy frecuentes (27) | 3 | |
| Hematuria | Muy frecuentes (10) | <1 | |
| Epistaxis | Frecuentes (7) | <1 | |
| Hemorragia gastrointestinal | Raras (<1) | <1 | |
| Hipertensión§ | Muy frecuentes (13) | 7 | |
| Trastornos respiratorios, torácicos y mediastínicos | Tos | Muy frecuentes (19) | <1 |
| Trastornos gastrointestinales | Diarrea | Muy frecuentes (19) | 2 |
| Estreñimiento | Muy frecuentes (12) | <1 | |
| Trastornos de la piel y del tejido subcutáneo | Erupción cutánea§ | Muy frecuentes (23) | <1 |
| Prurito | Frecuentes (7) | <1 | |
| Dermatitis exfoliativa general | Frecuencia no conocida | Frecuencia no conocida | |
| Trastornos musculoesque-léticos y del tejido conjuntivo | Dolor musculoesquelético§ | Muy frecuentes (23) | 2 |
| Artralgia | Muy frecuentes (13) | <1 | |
| Dolor de espalda | Muy frecuentes (10) | <1 | |
| Trastornos generales y afecciones en el lugar de administración | Fatiga§ | Muy frecuentes (16) | 1 |
| Fatiga | Muy frecuentes (12) | 1 | |
| Astenia | Frecuentes (4) | <1 | |
| Edema periférico | Frecuentes (7) | <1 | |
| Trastornos del metabolismo y de la nutrición | Síndrome de lisis tumoral§# | Raras (<1) | <1 |
| Exploraciones complementarias | Recuento absoluto de neutrófilos disminuido†± | Muy frecuentes (49) | 21 |
| Plaquetas disminuidas†± | Muy frecuentes (36) | 7 | |
| Hemoglobina disminuida†± | Muy frecuentes (23) | 4 |
* Los grados se evaluaron según los criterios de terminología para acontecimientos adversos del National Cancer Institute Common terminology (NCI-CTCAE), versión 4.03.
† Basado en las mediciones analíticas
± Los porcentajes se basan en el número de pacientes que tenían evaluaciones iniciales y al menos una posterior al inicio
§ Incluye varios términos de reacciones adversas
# Incluye acontecimientos con desenlace mortal
Otra población especial
Pacientes de edad avanzada
De los 1550 pacientes tratados con BRUKINSA, el 61,3 % tenía 65 años o más. La incidencia de acontecimientos adversos de grado 3 o superior fue ligeramente mayor entre los pacientes de edad avanzada tratados con zanubrutinib (60,3 % de los pacientes ≥65 años frente a 54,0 % de los pacientes <65 años). No se observaron diferencias clínicamente relevantes en la seguridad entre los pacientes ≥65 años y más jóvenes.
Población pediátrica
No se ha establecido la seguridad y eficacia de BRUKINSA en niños y adolescentes menores de 18 años.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es
4.9 - Sobredosificación de BRUKINSA 80 mg Cáps. dura
No hay ningún antídoto específico para BRUKINSA. Los pacientes que sufran una sobredosis deben ser controlados de cerca y recibir el tratamiento sintomático adecuado.
5. - PROPIEDADES FARMACOLÓGICAS
5.1 - Propiedades farmacodinámicas de BRUKINSA 80 mg Cáps. dura
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores de la tirosina cinasa de Bruton, código ATC: L01EL03.
Mecanismo de acción
El zanubrutinib es un inhibidor de la tirosina cinasa de Bruton (BTK). El zanubrutinib forma una unión covalente con un residuo de cisteína en el sitio activo de la BTK, lo que da lugar a la inhibición de la actividad de la BTK. La BTK es una molécula de señalización del receptor antigénico de linfocitos B (RBQ) y las vías receptoras de citocinas. En los linfocitos B, la señalización de la BTK se traduce en la activación de las vías necesarias para la proliferación, el tráfico, la quimiotaxis y la adhesión de los linfocitos B.
Efectos farmacodinámicos
Ocupación de la BTK en CMSP y biopsias de ganglios linfáticos
La mediana de la ocupación de la BTK en equilibrio dinámico en células mononucleares de sangre periférica se mantuvo en el 100 % más de 24 horas a una dosis diaria total de 320 mg en pacientes con neoplasias malignas de células B. La mediana de la ocupación de la BTK en los ganglios linfáticos fue de entre el 94 % y el 100 % después de la dosis recomendada.
Efecto en el intervalo QT/QTc y la electrofisiología cardíaca
A la dosis recomendada (320 mg una vez al día o 160 mg dos veces al día), no hubo efectos clínicamente relevantes sobre el intervalo QTc. A una dosis única 1,5 veces la dosis máxima recomendada (480 mg), el zanubrutinib no prolongó el intervalo QT a ningún grado clínicamente relevante (es decir, ≥10 ms).
Eficacia y seguridad clínicos
Pacientes con macroglobulinemia de Waldenström (MW)
La seguridad y la eficacia de BRUKINSA en la MW se evaluaron en un estudio aleatorizado, abierto y multicéntrico que comparó el zanubrutinib y el ibrutinib (estudio ASPEN) en pacientes sin tratamiento previo con inhibidor de BTK. Los pacientes aptos tenían al menos 18 años de edad y un diagnóstico histológico clínico y confirmado de MW recidivante/resistente o sin tratamiento previo si el médico responsable del tratamiento los consideró inadecuados para los regímenes de quimioinmunoterapia habituales. Los pacientes tenían que cumplir al menos un criterio para el tratamiento de acuerdo con los criterios de consenso del Séptimo Grupo de Trabajo Internacional sobre Macroglobulinemia de Waldenström (IWWM) y tener enfermedad medible, definida por un nivel de IgM en suero >0,5 g/dl. Los pacientes con mutación MYD88 (MYD88MUT) fueron asignados a la cohorte 1 (N = 201) y aleatorizados 1:1 para recibir zanubrutinib 160 mg dos veces al día (grupo A) o ibrutinib 420 mg una vez al día (grupo B) hasta la progresión de la enfermedad o toxicidad inaceptable. Los sujetos que presentaban MYD88 de tipo natural (MYD88WT) en la secuenciación génica (según las estimaciones, aproximadamente el 10 % de los sujetos inscritos) fueron inscritos en la cohorte 2 (N = 28) y recibieron zanubrutinib 160 mg dos veces al día en un tercer grupo del estudio no aleatorizado (grupo C).
En la cohorte 1, (MYD88MUT), la mediana de edad fue de 70 años (intervalo, 38 a 90 años), siendo el 71 % y el 60 % de los pacientes tratados con ibrutinib y zanubrutinib, respectivamente, >65 años. El 33 % de los pacientes del grupo de zanubrutinib y el 22 % de los de ibrutinib tenía >75 años. El 67 % era de sexo masculino y el 91 % era de raza blanca. Al inicio del estudio, el 44 % de los pacientes del grupo de ibrutinib y el 46 % de los pacientes del grupo de zanubrutinib tenía una puntuación alta en el Sistema Internacional de Puntuación de Pronósticos (IPSS). Ciento sesenta y cuatro pacientes presentaban enfermedad recidivante o resistente; la mediana del número de tratamientos previos era de 1 (intervalo, de 1 a 8).
El criterio de valoración principal fue la tasa de respuesta completa (RC) o respuesta parcial muy buena (RPMB), evaluada por un comité de revisión independiente (CRI) con la adaptación de los criterios de respuesta actualizados en el Sexto IWWM. Los criterios de valoración secundarios para la cohorte 1 son la tasa de respuesta mayor (TRM), la duración de la respuesta, la tasa de RC o RPMB determinada por el investigador y la supervivencia sin progresión (SSP).
El análisis de la superioridad del criterio de valoración principal de la tasa de RPMB o RC precisó análisis en el conjunto de análisis con enfermedad recidivante/resistente antes del análisis en el conjunto de análisis por IT. La mediana del seguimiento fue de 19,4 meses.
En los pacientes con enfermedad recidivante/resistente, el 19,8 % y el 28,9 % alcanzaron RPMB o RC en los grupos de ibrutinib y zanubrutinib, respectivamente. El criterio de valoración principal de la eficacia no fue significativo en el conjunto de análisis con enfermedad recidivante/resistente (p bilateral = 0,1160). En la Tabla 4 se resumen las respuestas evaluadas por el CRI para el conjunto de análisis con enfermedad recidivante/resistente y por IT. Se observaron respuestas con zanubrutinib en los subgrupos, incluidos los pacientes con MYD88WT (cohorte 2), que presentaron una RPMB o una RC del 26,9 % y una TRM del 50 %.
Tabla 4: Análisis principal de la respuesta de la enfermedad según un comité de revisión independiente (estudio ASPEN)
| Categoría de respuesta | Recidivante/resistente | ITT | ||
| Ibrutinib N = 81 | Zanubrutinib N = 83 | Ibrutinib N = 99 | Zanubrutinib N = 102 | |
| Mediana del tiempo de seguimiento, meses (intervalo) | 18,79 (0,5, 30,0) | 18,73 (0,4, 28,7) | 19,38 (0,5, 31,1) | 19,47 (0,4, 31,2) |
| RC | 0 (0,) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
| RPMB | 16 (19,8) | 24 (28,9) | 19 (19,2) | 29 (28,4) |
| RP | 49 (60,5) | 41 (49,4) | 58 (58,6) | 50 (49,0) |
| Tasa de RPMB o RC, n (%) | 16 (19,8) | 24 (28,9) | 19 (19,2) | 29 (28,4) |
| IC del 95 %a | (11,7, 30,1) | (19,5, 39,9) | (12,0, 28,3) | (19,9, 38,2) |
| Diferencia de riesgos (%)b | 10,7 | 10,2 | ||
| IC del 95 %a | (-2,5, 23,9) | (-1,5, 22,0) | ||
| Valor de pc | 0,1160 | |||
| TRM (RP o mejor), n (%) | 65 (80,2) | 65 (78,3) | 77 (77,8) | 79 (77,5) |
| IC del 95 %a | (69,9, 88,3) | (67,9, 86,6) | (68,3, 85,5) | (68,1, 85,1) |
| Diferencia de riesgos (%)b | -3.5 | -0,5 | ||
| IC del 95 % | (-16,0, 9,0) | (-12,2, 11,1) | ||
| Duración de la respuesta mayor | ||||
| Tasa sin acontecimientos, % (IC del 95 %)d 18 meses | 85,6 (73,1, 92,6) | 87,0 (72,5, 94,1) | 87,9 (77,0, 93,8) | 85,2 (71,7, 92,6) |
Los porcentajes se basan en N.
a Intervalo de confianza del 95 % bilateral de Clopper-Pearson.
b Diferencia del riesgo común de Mantel-Haenszel con el intervalo de confianza del 95 % calculado mediante la aproximación normal y el error estándar de Sato mediante los factores de estratificación por el IRT (los estratos de CXCR4 WT y DESC están combinados) y el grupo de edad (≤65 y >65). El ibrutinib es el grupo de referencia.
c Basado en la prueba de CMH estratificada según los factores de estratificación por el IRT (los estratos de CXCR4 WT y DESC están combinados) y el grupo de edad (≤65 y >65)
d Las tasas sin acontecimientos se calculan mediante el método de Kaplan-Meier con IC del 95 % calculados mediante la fórmula de Greenwood.
A partir de un valor de corte de los datos actualizado, la tasa sin acontecimientos en la supervivencia sin progresión según la evaluación del investigador fue del 77,6 % frente al 84,9 % a los 30 meses (ibrutinib frente a zanubrutinib), con un cociente de riesgos instantáneos estimado de 0,734 (IC del 95 %: 0,380, 1,415).
Pacientes con linfoma de la zona marginal (LZM)
La eficacia de zanubrutinib se evaluó en un ensayo de fase II, abierto, multicéntrico y de un solo grupo de 68 pacientes con LZM que habían recibido al menos un tratamiento previo con anticuerpos anti-CD20. Veintiséis (38,2 %) pacientes presentaban LZM extraganglionar, 26 (38,2 %) LZM ganglionar, 12 (17,6 %) LZM esplénico y en 4 (6 %) pacientes se desconocía el subtipo. Se administró zanubrutinib por vía oral en una dosis de 160 mg dos veces al día hasta la progresión de la enfermedad o toxicidad inaceptable. La mediana de edad de los pacientes era de 70 años (intervalo, de 37 a 95), y el 53 % eran varones. La mediana del tiempo transcurrido desde el diagnóstico inicial fue de 61,5 meses (intervalo, de 2,0 a 353,6). La mediana del número de tratamientos previos era de 2 (intervalo, de 1 a 6), y el 27,9 % de los pacientes habían recibido 3 líneas o más de terapia sistémica; el 98,5 % (n = 67) de los pacientes habían recibido quimioterapia con rituximab previa y el 85,3 % (n = 58) de los pacientes habían recibido tratamiento previo con fármacos alquilantes; el 5,9 % de los pacientes (n = 4) habían recibido un trasplante de células madre previo. Sesenta y tres (92,6 %) pacientes tenían un estado funcional ECOG inicial de 0 o 1. Veintidós (32,4 %) pacientes mostraban enfermedad resistente al inicio del estudio.
La respuesta tumoral se evaluó según la clasificación de Lugano de 2014, y la variable principal de eficacia fue la tasa de respuesta global evaluada por un comité de revisión independiente (CRI).
Tabla 3. Resultados de eficacia en pacientes con LZM según un comité de revisión independiente
| Estudio BGB-3111-214 (N = 66)* | |
| TRG (IC del 95 %) | 68 % (55,6; 79,1) |
| RC | 26 % |
| RP | 42 % |
| Mediana de la DdR en meses (IC del 95 %) | NE (25,0; NE) |
| Tasa de DdR libre de acontecimientos b a los 24 meses, % (IC del 95 %) | 72,9 (54,4; 84,9) |
| Mediana del seguimiento del estudio en meses (mín., máx.) | 28,04 (1,64; 32,89) |
a No se pudo evaluar la eficacia en dos pacientes del estudio BGB-3111-214 debido a la confirmación central de transformación del LZM en linfoma difuso de células B grandes.
b Las tasas libres de acontecimientos se calcularon con el método de Kaplan-Meier y los IC del 95 % correspondientes, con la fórmula de Greenwood.
TRG: tasa de respuesta global, RC: respuesta completa, RP: respuesta parcial, DdR: duración de la respuesta, IC: intervalo de confianza, NE: no estimable.
En el estudio BGB-3111-214, la mediana del tiempo transcurrido hasta la respuesta fue de 2,79 meses (intervalo, de 1,7 a 11,1 meses). Después de un tiempo de seguimiento de 28,04 meses de mediana (intervalo, de 1,64 a 32,89 meses), la mediana de la duración de la respuesta (DdR) evaluada por el CRI no se había alcanzado (IC del 95 %, de 25,0 meses a NE) y se estimó que en total el 72,9 % (IC del 95 %, de 54,4 a 84,9) de los pacientes con respuesta estaban libres de acontecimientos 24 meses después de la respuesta inicial.
Las tasas de respuesta global observadas fueron similares en los tres tipos diferentes de LZM (extraganglionar, ganglionar y esplénico).
Pacientes con leucemia linfocítica crónica (LLC)
La eficacia de BRUKINSA en los pacientes con LLC se evaluó en dos ensayos controlados aleatorizados.
BGB-3111-304: Estudio internacional, de fase III, abierto y aleatorizado de zanubrutinib comparado con bendamustina más rituximab (BR) en pacientes con LLC no tratada previamente.
BGB-3111-304 es un ensayo de fase III aleatorizado, multicéntrico y abierto con control activo de zanubrutinib en monoterapia y bendamustina en combinación con rituximab en 479 pacientes con LLC no tratada previamente sin deleción 17p (del(17p)) (grupos A y B, cohorte 1). El grupo C de BGB-3111-304 (cohorte 2) es un ensayo multicéntrico de un solo grupo de zanubrutinib en monoterapia en 110 pacientes con LLC no tratada previamente con del(17p) confirmada centralmente.
Ambas cohortes incluyeron pacientes de 65 años o más, así como pacientes de entre 18 y 65 años que no eran aptos para quimioinmunoterapia con fludarabina, ciclofosfamida y rituximab (FCR).
Por lo general, las características demográficas e iniciales estaban bien equilibradas entre el grupo A (zanubrutinib) y el grupo B (BR) de la cohorte 1. En ambos grupos, la mediana de la edad se situó en 70,0 años, con una proporción ligeramente superior de pacientes ≥75 años (26,1 %) en el grupo A en comparación con el grupo B (22,3 %) y una proporción ligeramente inferior de pacientes de 65-75 años (55,2 %) en el grupo A en comparación con el grupo B (58,4 %). En la cohorte 1, el 92,7 % de los pacientes tenían un estado funcional ECOG inicial de 0 o 1 (93,7 % en el grupo A y 91,6 % en el grupo B). En la cohorte 2 (grupo C de zanubrutinib), el 87,3 % de los pacientes tenían un estado funcional ECOG de 0 o 1.
Por lo general, las características demográficas e iniciales también estaban bien equilibradas entre el grupo A (zanubrutinib) y el grupo C (zanubrutinib) de la cohorte 2.
En la cohorte 1, la aleatorización se estratificó por edad (<65 edad frente a ≥65 años), el estadio de Binet (C frente a A o B), el estado mutacional de la región variable de la cadena pesada de la inmunoglobulina (IGHV) (mutada frente a no mutada) y región geográfica (América del Norte frente a Europa frente a Asia-Pacífico). Fueron aleatorizados 479 pacientes en total (grupo de análisis por intención de tratar [ITT]), 241 a monoterapia continua con zanubrutinib y 238 a 6 ciclos de tratamiento con bendamustina y rituximab (BR).
En cohorte 1, los pacientes del grupo A de zanubrutinib recibieron 160 mg dos veces al día hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. En el grupo B, los pacientes recibieron bendamustina a una dosis de 90 mg/m2/día los 2 primeros días de cada ciclo durante 6 ciclos y rituximab a una dosis de 375 mg/m2 durante el día 1, y a una dosis de 500 mg/m2 durante los ciclos 2-6. Cada ciclo de tratamiento consistió en 28 días aproximadamente. En la cohorte 2 (grupo C), los pacientes recibieron 160 mg de zanubrutinib dos veces al día hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable.
En la cohorte 1, el criterio de valoración principal fue la supervivencia sin progresión (SSP), evaluada por un comité de revisión independiente (CRI). Los criterios de valoración secundarios incluyeron la tasa de respuesta global según la evaluación del CRI.
En la cohorte 1, la mediana de la duración del seguimiento de la SSP fue de 25,0 meses (intervalo: de 0,0 a 41,4). La tasa de SSP a los 24 meses se situó en el 85,5 % (IC del 95 %: 80,1 89,6) con zanubrutinib y en el 69,5 % (IC del 95 %: 62,4, 75,5) con BR. En la cohorte 2, la mediana de la duración del seguimiento de la SSP fue de 27,9 meses (intervalo: de 1,0 a 38,8) y la tasa de SSP a los 24 meses, de 88,9 % (IC del 95 %: 81,3, 93,6). La TRG evaluada por el CRI en la cohorte 2 fue del 90,0 % (IC del 95 %: 82,8, 94,9). La mediana del tiempo hasta una respuesta parcial o superior evaluada por el CRI fue de 2,89 meses (intervalo: 1,8, 14,2) y 2,86 meses (intervalo: 1,9, 13,9) en el grupo de zanubrutinib de la cohorte 1 y la cohorte 2, respectivamente.
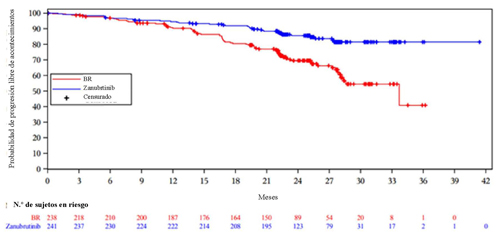
Los resultados de la eficacia de la cohorte 1 se presentan en la Tabla . Las curvas de Kaplan-Meier de la SSP de ambos grupos de la cohorte 1 se muestran en la Figura 1.
Tabla 6: Resultados de la eficacia en BGB-3111-304
| Cohorte 1* Pacientes sin del(17p) | ||
| Criterio de valoración | Zanubrutinib (N = 241) | Bendamustina + Rituximab (N = 238) |
| Supervivencia sin progresión | ||
| Número de acontecimientos, n (%) | 36 (14,9) | 71 (29,8) |
| Progresión de la enfermedad, n (%) | 27 (11,2) | 59 (24,8) |
| Muerte, n (%) | 9 (3,7) | 12 (5,0) |
| Mediana (IC del 95 %), meses a | NE (NE, NE) | 33,7 (28,1, NE) |
| Cociente de riesgos instantáneos (IC del 95 %) b | 0,42 (0,28, 0,63) | |
| Valor de P c | <0,0001 | |
| Tasa de respuesta global† % (IC del 95 %) | 94,6 % (91,0, 97,1) | 85,3 % (80,1, 89,5) |
Tasa de respuesta global: RC+RCi+RPg+RP+RP-L, RC: respuesta completa, RCi: respuesta completa con recuperación hematopoyética incompleta, RPg: respuesta parcial ganglionar, RP: respuesta parcial, RP-L: respuesta parcial con linfocitoma, IC: intervalo de confianza, NE: no estimable, la mediana del tiempo de seguimiento de la SSP fue de 25,0 meses (IC del 95 %: 24,6, 25,2).
* Grupo de análisis ITT
† Evaluado por el comité de revisión central independiente.
a Basado en la estimación de Kaplan-Meier.
b Basado en un modelo de regresión de Cox estratificado con bendamustina + rituximab como grupo de referencia.
c Basado en una prueba de rangos logarítmicos estratificada.
En un análisis ad hoc actualizado con una mediana de seguimiento de la SSP de 33,5 meses, la SSP evaluada por el investigador se mantuvo coherente con el análisis principal, con un CRI de 0,33 (IC del 95 %: de 0,22 a 0,48, P descriptiva < 0,0001) en el grupo de zanubrutinib con respecto al de BR. En el grupo de zanubrutinib no se alcanzó la mediana de la SSP, y esta fue de 39,2 meses en el grupo de BR. A los 36 meses de la aleatorización, se estimó que el 83,6 % de los pacientes tratados con zanubrutinib y el 55,1 % de los que recibieron BR permanecían sin progresión y seguían vivos. Con una mediana de seguimiento de 35,8 meses, no se alcanzó la mediana de la SG en ninguno de los dos grupos; la estimación de la tasa de SG a los 36 meses fue del 90,9 % (IC del 95 %: de 86,3 a 94,0) en el grupo de zanubrutinib, y del 89,5 % (IC del 95 %: de 84,2 a 93,1) en el grupo de BR, respectivamente.
Figura 1: Curva de Kaplan-Meier de la SSP evaluada por el CRI en la cohorte 1 de BGB-3111-304 (población ITT)
BGB-3111-305: Estudio de fase III aleatorizado de zanubrutinib comparado con ibrutinib en pacientes con LLC recidivante/resistente (R/R)
BGB-3111-305 es un estudio de fase III aleatorizado, multicéntrico, abierto y con control activo. Incluyó a 652 pacientes con LLC recidivante o resistente después de al menos un tratamiento sistémico previo. Los pacientes fueron aleatorizados a 160 mg de zanubrutinib por vía oral dos veces al día o a 420 mg de ibrutinib por vía oral una vez al día, que se administraron de forma continua hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable.
La aleatorización se estratificó por edad (<65 años frente a ≥65 años), región geográfica (China frente a fuera de China), estado resistente (sí o no) y estado mutacional del(17p)/TP53 (presente o ausente).
Por lo general, las características demográficas e iniciales estaban bien equilibradas entre los grupos de tratamiento del grupo de análisis ITT y los 415 primeros pacientes aleatorizados.
En el grupo de análisis ITT, la mediana de la edad se situó en 67,0 años en el grupo de zanubrutinib y en 68,0 años en el grupo de ibrutinib. La mayoría de los pacientes de ambos grupos tenía un EF ECOG de 0 o 1 (97,9 % en el grupo de zanubrutinib; 96,0 % en el grupo de ibrutinib). En los 415 primeros pacientes aleatorizados se observaron unas características demográficas e iniciales similares. La mediana del número de líneas anteriores de tratamiento sistémico es de 1,0 en el grupo de zanubrutinib (intervalo, de 1 a 6) y de 1,0 en el grupo de ibrutinib (intervalo, de 1 a 8) tanto en el grupo de análisis ITT como en los 415 primeros pacientes aleatorizados.
Los pacientes tratados previamente con un inhibidor de la BTK fueron excluidos del estudio 305 y los datos disponibles de zanubrutinib después de un tratamiento de inhibición de la BTK son limitados.
De los 652 pacientes totales, 327 fueron asignados a zanubrutinib en monoterapia y 325 a ibrutinib en monoterapia. La evaluación de la eficacia se basa en el análisis provisional preespecificado de los 415 primeros pacientes aleatorizados de la población ITT. De estos, 207 fueron aleatorizados a zanubrutinib en monoterapia y 208 a ibrutinib en monoterapia. Los resultados de la eficacia se presentan en la tabla 7.
El criterio de valoración principal fue la tasa de respuesta global (TRG, definida como respuesta parcial o mejor).
En el análisis provisional preespecificado de la TRG en los 415 pacientes aleatorizados, zanubrutinib demostró no inferioridad (p unilateral <0,0001) y superioridad (p bilateral = 0,0006) con respecto al ibrutinib en el criterio de valoración principal preespecificado del protocolo de la TRG evaluada por el investigador. La respuesta determinada por el CRI también demostró la no inferioridad de zanubrutinib con respecto al ibrutinib (p unilateral <0,0001). En el análisis final de la TRG, la TRG evaluada por el investigador continúa siendo superior (79,5 % frente a 71,1 %) en el grupo de zanubrutinib en comparación con el grupo de ibrutinib (p descriptiva = 0,0133); la TRG determinada por el CRI también fue significativamente superior en el grupo de zanubrutinib que en el de ibrutinib, lo que demuestra su superioridad (80,4 % frente a 72,9 % respectivamente; p bilateral = 0,0264).
Tabla 7: Resultados de la eficacia en BGB-3111-305 (análisis final de los primeros 415 pacientes aleatorizados) según las evaluaciones del investigador (criterio de valoración principal preespecificado del protocolo) y del CRI
| Evaluado por el investigador (criterio de valoración principal preespecificado del protocolo) | Evaluado por el CRI | |||
| Criterio de valoración | Zanubrutinib (N = 207) | Ibrutinib (N = 208) | Zanubrutinib (N = 207) | Ibrutinib (N = 208) |
| (IC del 95 %) | (72,0, 83,7) | (55,5, 69,1) | (69,9, 81,9) | (57,5, 70,9) |
| Cociente de repuesta a (IC del 95 %) | 1,25 (1,10, 1,41) | 1,17 (1,04, 1,33) | ||
| No inferioridad b | Valor de p unilateral <0,0001 | Valor de p unilateral <0,0001 | ||
| Superioridad c | Valor de p bilateral 0,0006 | Valor de p bilateral 0,0121 | ||
| Duración de la respuesta d: Tasa de ausencia de acontecimientos a los 12 meses % (IC del 95 %) | 89,8 (78,1, 95,4) | 77,9 (64,7, 86,7) | 90,3 (82,3, 94,8) | 78,0 (66,1, 86,2) |
Tasa de respuesta global: RC+RCi+RPg+RP, RC: respuesta completa, RCi: respuesta completa con recuperación hematopoyética incompleta, RPg: respuesta parcial ganglionar, RP: respuesta parcial, IC: intervalo de confianza La mediana de la duración de la respuesta evaluada por el investigador no se alcanzó en el grupo de zanubrutinib en el análisis final; la mediana del tiempo del seguimiento del estudio fue de 15,31 meses (intervalo: 0,1, 23,1) en el grupo de zanubrutinib y de 15,43 meses (intervalo: 0,1, 26,0) en el grupo de ibrutinib.
§ Las pruebas de la hipótesis de no inferioridad de la TRG en el análisis provisional se basan en los primeros 415 pacientes aleatorizados solo a un nivel de significación unilateral de 0,005.
a Cociente de respuesta: cociente estimado de la tasa de respuesta global en el grupo de zanubrutinib dividido por la del grupo de ibrutinib.
b Prueba estratificada con un cociente de respuesta nula de 0,8558.
c Prueba de Cochran-Mantel-Haenszel estratificada.
d Estimación de Kaplan-Meier.
La mediana del tiempo hasta la respuesta evaluada por el investigador en el análisis provisional de la TRG de los 415 primeros pacientes aleatorizados fue de 5,59 meses (intervalo: 2,7, 14,1) en el grupo de zanubrutinib y de 5,65 meses (intervalo: 2,8, 16,7) en el grupo de ibrutinib. Los resultados evaluados por el CRI fueron coherentes (5,55 meses frente a 5,63 meses en los grupos de zanubrutinib e ibrutinib, respectivamente). En el análisis final de la TRG de los 652 pacientes aleatorizados, la mediana del tiempo hasta la respuesta permaneció inalterada (5,59 meses frente a 5,65 meses según la evaluación del investigador y 5,52 meses frente a 5,62 meses según la evaluación del CRI en los grupos de zanubrutinib e ibrutinib, respectivamente).
En los 415 primeros pacientes, en los pacientes con mutación del(17p) la TRG evaluada por el investigador fue del 83,3 % (IC del 95 % 62,5, 95,3; 20 de 24 pacientes) en el grupo de zanubrutinib y del 53,8 % (IC del 95 % 33,4, 73,4; 14 de 26 pacientes) en el grupo de ibrutinib. Según la evaluación del CRI, la TRG se situó en el 79,2 % (IC del 95 % 57,8, 92,9; 19 de 24 pacientes) en el grupo de zanubrutinib y en el 61,5 % (IC del 95 % 40,6, 79,8; 16 de 26 pacientes) en el grupo de ibrutinib. En el análisis final de la TRG en los 652 pacientes aleatorizados, la TRG evaluada por el investigador se situó en el 86,7 % (IC del 95 % 73,2, 94,9; 39 de 45 pacientes con mutación del(17p)) en el grupo de zanubrutinib y 56,0 % (IC del 95 % 41,3, 70,0; 28 de 50 pacientes con mutación del(17p)) en el grupo de ibrutinib. Según la evaluación del CRI, la TRG se situó en el 86,7 % (IC del 95 % 73,2, 94,9; 39 de 45 pacientes con mutación del(17p)) en el grupo de zanubrutinib y en el 64,0 % (IC del 95 % 49,2, 77,1; 32 de 50 pacientes con mutación del(17p)) en el grupo de ibrutinib.
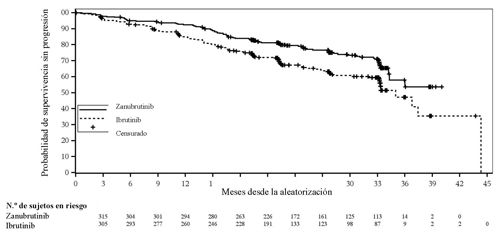
En el momento preespecificado para el análisis final de la SSP (fecha de corte: 8 de agosto de 2022) se había incluido a un total de 652 pacientes. La mediana del tiempo de seguimiento de la SSP era de 28,1 meses según la evaluación del investigador, y de 30,7 meses según el CRI. Zanubrutinib mostró superioridad en cuanto a la SSP con respecto a ibrutinib según la evaluación del investigador y del CRI. Los resultados de eficacia de la SSP se presentan en la Tabla 8, y en la Figura 2 se ofrece una curva de Kaplan-Meier de la evaluación efectuada por el CRI.
Tabla 8. Resultados de eficacia en BGB-3111-305 (análisis final de la SSP preespecificado de los 652 pacientes aleatorizados) según las evaluaciones del investigador y del CRI (fecha de corte: 8 de agosto de 2022)
| Variable | Evaluación del investigador | Evaluación independiente* | ||
| Zanubrutinib (N = 327) | Ibrutinib (N = 325) | Zanubrutinib (N = 327) | Ibrutinib (N = 325) | |
| Supervivencia sin progresión | ||||
| Acontecimientos, n (%) | 87 (26,6) | 118 (36,3) | 88 (26,9) | 120 (36,9) |
| Cociente de riesgos instantáneosa (IC del 95 %) | 0,65 (0,49, 0,86) | 0,65 (0,49, 0,86) | ||
| Valor de p bilateralb | 0,0024 | 0,0024 | ||
a Según un modelo de regresión de Cox estratificado con ibrutinib como grupo de referencia.
b Según una prueba de rangos logarítmicos estratificada.
Figura 2. Curva de Kaplan-Meier de la supervivencia sin progresión según una revisión central independiente (ITT) (fecha de corte: 8 de agosto de 2022)
En los pacientes con la mutación del(17p)/TP53, el cociente de riesgos instantáneos de la supervivencia sin progresión según la evaluación del investigador fue de 0,53 (IC del 95 % 0,31, 0,88). En una revisión independiente, el cociente de riesgos instantáneos fue de 0,52 (IC del 95 % 0,30, 0,88) (Figura 3).
Figura 3: Curva de Kaplan-Meier de la supervivencia sin progresión según una revisión central independiente de los pacientes con Del 17P o TP53 (ITT) (fecha de corte: 8 de agosto de 2022)
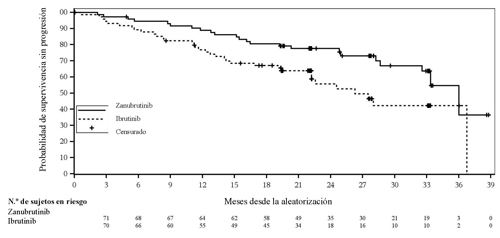
Con una mediana de seguimiento estimada de 32,8 meses, no se alcanzó la mediana de supervivencia general en ninguno de los dos grupos; el 17 % de los pacientes experimentaron algún acontecimiento.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con BRUKINSA en todos los grupos de la población pediátrica para el tratamiento del linfoma linfoplasmocítico y para el tratamiento de las neoplasias de células B maduras (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 - Propiedades farmacocinéticas de BRUKINSA 80 mg Cáps. dura
La concentración plasmática máxima (Cmáx) de zanubrutinib y el área bajo la curva de la concentración plasmática del fármaco a lo largo del tiempo (AUC) aumentan de manera proporcional más de un intervalo de dosis de 40 mg a 320 mg (de 0,13 a 1 vez la dosis diaria total recomendada). Se ha observado una acumulación sistémica limitada de zanubrutinib después de la administración repetida durante una semana.
La media geométrica (%CV) del AUC diaria en equilibrio dinámico del zanubrutinib es 2,099 (42 %) ng·h/ml después de 160 mg dos veces al día y de 1,917 (59 %) ng·h/ml después de 320 mg una vez al día. La media geométrica (%CV) de la Cmáx en equilibrio dinámico del zanubrutinib es 299 (56 %) ng/ml después de 160 mg dos veces al día y de 533 (55 %) ng/ml después de 320 mg una vez al día.
Absorción
La mediana del tmáx del zanubrutinib es de 2 horas. No se observaron diferencias clínicamente significativas en el AUC o la Cmáx del zanubrutinib después de la administración de una comida rica en grasas (aproximadamente 1000 calorías con el 50 % de contenido de calorías totales procedentes de grasas) en sujetos sanos.
Distribución
La media geométrica (%CV) del volumen de distribución aparente en equilibrio dinámico del zanubrutinib durante la fase terminal (Vz/F) fue de 522 l (71 %). La fijación a proteínas plasmáticas del zanubrutinib es del 94 % aproximadamente y el cociente de sangre y plasma, de 0,7-0,8.
Metabolismo
El zanubrutinib se metaboliza principalmente mediante el citocromo P450(CYP)3A.
Eliminación
La semivida media (t½) del zanubrutinib es de aproximadamente entre 2 y 4 horas después de una única dosis oral de 160 mg o 320 mg de zanubrutinib. La media geométrica (%CV) del aclaramiento oral aparente (CL/F) del zanubrutinib durante la fase terminal fue de 128 (61 %) l/h. Después de una única dosis de 320 mg zanubrutinib radiomarcado a sujetos sanos, aproximadamente el 87 % de la dosis se recuperó en las heces (38 % inalterado) y el 8 % en la orina (menos del 1 % inalterado).
Poblaciones especiales
Pacientes de edad avanzada
La edad (de 19 a 90 años; media de edad 65 ± 12,5) no tuvo ningún efecto clínicamente relevante sobre la farmacocinética del zanubrutinib según el análisis FC poblacional (N = 1291).
Población pediátrica
No se han realizado estudios farmacocinéticos con zanubrutinib en pacientes menores de 18 años de edad.
Sexo
El sexo (872 varones y 419 mujeres) no tuvo ningún efecto clínicamente relevante sobre la farmacocinética del zanubrutinib según el análisis FC poblacional.
Raza
La raza (964 de raza blanca, 237 oriental, 30 negra y 25 de otras) no tuvo ningún efecto clínicamente relevante sobre la farmacocinética del zanubrutinib según el análisis FC poblacional.
Peso corporal
El peso corporal (de 36 a 149 kg, peso medio de 76,5 ± 16,9 kg) no tuvo ningún efecto clínicamente relevante sobre la farmacocinética del zanubrutinib según el análisis FC poblacional (N = 1291).
Insuficiencia renal
El zanubrutinib es objeto de una eliminación renal mínima. Según el análisis FC poblacional, la insuficiencia renal leve y moderada (aclaramiento de creatinina [ClCr] ≥30 ml/min calculado con la ecuación de Cockcroft-Gault) no influyó en la exposición al zanubrutinib. El análisis se basó en 362 pacientes con función renal normal, 523 con insuficiencia renal leve, 303 con insuficiencia renal moderada, 11 con insuficiencia renal grave y uno con ESRD. Se desconocen los efectos de la insuficiencia renal grave (CrCl <30 ml/min) y la diálisis en la farmacocinética del zanubrutinib.
Insuficiencia hepática
El AUC total del zanubrutinib aumentó el 11 % en los sujetos con insuficiencia hepática leve (Child-Pugh de clase A), en el 21 % en los sujetos con insuficiencia hepática moderada (clase B de Child-Pugh) y en el 60 % en los sujetos con insuficiencia hepática grave (clase C de Child-Pugh) en relación con los pacientes con función hepática normal. El AUC libre del zanubrutinib aumentó el 23 % en los sujetos con insuficiencia hepática leve (Child-Pugh de clase A), en el 43 % en los sujetos con insuficiencia hepática moderada (clase B de Child-Pugh) y en el 194 % en los sujetos con insuficiencia hepática grave (clase C de Child-Pugh) en relación con los pacientes con función hepática normal. Se observó una correlación significativa entre la puntuación de Child-Pugh, el valor inicial de albúmina sérica, el valor inicial de bilirrubina sérica y el tiempo de protrombina inicial con AUC de zanubrutinib libre.
Estudios in vitro
Enzimas de CYP
Zanubrutinib es un inductor débil de CYP2B6 y CYP2C8. Zanubrutinib no es un inductor de CYP1A2.
Administración conjunta con sustratos/inhibidores transportadores
Zanubrutinib puede ser un sustrato de P-gp. Zanubrutinib no es un sustrato ni inhibidor de OAT1, OAT3, OCT2, OATP1B1 ni OATP1B3.
Interacciones farmacodinámicas
En un estudio in vitro se demostró que la posible interacción farmacodinámica entre zanubrutinib y rituximab es baja y es improbable que zanubrutinib interfiera en el efecto de citotoxididad celular dependiente de anticuerpos (ADCC) inducido por el anticuerpo anti-CD20.
Los estudios in vitro, ex vivo y con animales demostraron que zanubrutinib tuvo efectos nulos o insignificantes en la activación plaquetaria, la expresión de glicoproteínas y la formación de trombos.
5.3 - Datos preclínicos sobre seguridad de BRUKINSA 80 mg Cáps. dura
Toxicidad general
Los perfiles toxicológicos generales se zanubrutinib se caracterizaron de forma oral en ratas Sprague-Dawley durante un tratamiento de hasta 6 meses y en perros Beagle durante un tratamiento de hasta 9 meses.
En estudios de dosis repetidas en ratas hasta 6 meses de tratamiento, se observó mortalidad relacionada con el principio activo a la dosis de 1000 mg/kg/día (81 veces el AUC clínico) con hallazgos histopatológicos en el tubo gastrointestinal. Se observaron otros hallazgos principalmente en el páncreas (atrofia, fibroplasia, hemorragia y/o infiltración celular inflamatoria) a las dosis ≥30 mg/kg/día (3 veces el AUC clínico), en la piel alrededor de la nariz/boca/ojos (infiltración celular inflamatoria, erosión/úlcera) a partir de la dosis de 300 mg/kg/día (16x AUC clínica), y en el pulmón (presencia de macrófagos en el alveolo) a la dosis de 300 mg/kg/día. Todos estos hallazgos cambiaron total o parcialmente tras una recuperación de 6 semanas, excepto los hallazgos pancreáticos que no se consideraron clínicamente relevantes.
En los estudios de dosis repetidas en perros hasta los 9 meses de tratamiento, los hallazgos relacionados con el artículo de prueba se observaron principalmente en el tubo gastrointestinal (heces blandas/líquidas/mucosas), en la piel (erupción, coloración roja y engrosamiento/descamación) ) y en los ganglios linfáticos mesentéricos, mandibulares y asociados al intestino y al bazo (depleción linfoide o eritrofagocitosis) con las dosis de 10 mg/kg/día (3 veces el AUC clínico) a 100 mg/kg/día (18 veces el AUC clínico). Todos estos hallazgos cambiaron total o parcialmente tras una recuperación de 6 semanas.
Carcinogenia/genotoxicidad
No se han realizado estudios de carcinogenia con zanubrutinib.
El zanubrutinib no fue mutagénico en un ensayo de mutagenia bacteriana (Ames), no fue clastogénico en un ensayo de aberración cromosómica en células de mamíferos (CHO), ni fue clastogénico en un ensayo de micronúcleos de médula ósea in vivo en ratas.
Toxicidad reproductiva y durante el desarrollo’
Se llevó a cabo un estudio de fertilidad masculina y femenina y de desarrollo embrionario combinado en ratas con dosis orales de zanubrutinib de 30, 100 y 300 mg/kg/día. No se observaron efectos en la fertilidad masculina o femenina, pero en la dosis más alta analizada, se hallaron anomalías morfológicas en el esperma y aumento de las pérdidas posteriores a la implantación. La dosis de 100 mg/kg/día es aproximadamente 13 veces superior al de la exposición terapéutica en humanos.
Se realizaron estudios de toxicidad para el desarrollo embriofetal en ratas y conejos. El zanubrutinib se administró por vía oral a ratas preñadas durante el periodo de organogenia a las dosis de 30, 75, y 150 mg/kg/día. Se observaron malformaciones cardíacas (corazones con 2 o 3 cavidades a una incidencia del 0,3 %-1,5 %) a todos los niveles de dosis en ausencia de toxicidad materna. La dosis de 30 mg/kg/día es aproximadamente 5 veces superior a la exposición terapéutica en humanos.
La administración de zanubrutinib a conejas preñadas durante el periodo de organogénesis a 30, 70, y 150 mg/kg/día dio lugar a pérdida posterior a la implantación a la dosis más alta. La dosis de 70 mg/kg es de aproximadamente 25 veces superior a la exposición terapéutica en humanos y se asoció a toxicidad materna.
En un estudio de toxicidad para el desarrollo prenatal y posnatal, se administró zanubrutinib por vía oral a ratas a dosis de 30, 75 y 150 mg/kg/día desde la implantación hasta el destete. Las crías de los grupos de dosis medias y altas mostraron pesos corporales reducidos antes del destete, y todos los grupos de dosis presentaron hallazgos oculares adversos (p. ej., cataratas, protrusión del globo ocular). La dosis de 30 mg/kg/día es aproximadamente 5 veces superior a la exposición terapéutica en humanos.
6. - DATOS FARMACÉUTICOS
6.1 - Lista de excipientes de BRUKINSA 80 mg Cáps. dura
Contenido de las cápsulas
Celulosa microcristalina
Croscarmelosa de sodio
Lauril sulfato de sodio (E487)
Sílice, coloidal anhidro
Estearato de magnesio
Cuerpo de las cápsulas
Gelatina
Dióxido de titanio (E171)
Tinta de impresión
Goma laca (E904)
Óxido de hierro negro (E172)
Propilenglicol (E1520)
6.2 - Incompatibilidades de BRUKINSA 80 mg Cáps. dura
No procede.
6.3 - Período de validez de BRUKINSA 80 mg Cáps. dura
3 años.
6.4 - Precauciones especiales de conservación de BRUKINSA 80 mg Cáps. dura
No requiere condiciones especiales de conservación.
6.5 - Naturaleza y contenido del recipiente de BRUKINSA 80 mg Cáps. dura
Frascos de HDPE con cierre de seguridad para niños de polipropileno. Cada frasco contiene 120 cápsulas duras.
6.6 - Precauciones especiales de eliminación y otras manipulaciones de BRUKINSA 80 mg Cáps. dura
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. - TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
BeiGene Ireland Limited.
10 Earlsfort Terrace
Dublín 2
D02 T380
Irlanda
Tel. +353 1 566 7660
E-mail [email protected]
8. - NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/21/1576/001
9. - FECHA DE LA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
22 de noviembre de 2021
10. - FECHA DE LA REVISIÓN DEL TEXTO
25 de mayo de 2023
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.

