Informe de Posicionamiento Terapéutico de ibrutinib (Imbruvica®) en macroglobulinemia de Waldenström
INFORME DE POSICIONAMIENTO TERAPÉUTICO PT-IBRUTINIB_MW/V1/14042016
Informe de Posicionamiento Terapéutico de ibrutinib (Imbruvica®) en macroglobulinemia de Waldenström
Fecha de publicación: 14 de abril de 2016
RESUMEN
Ibrutinib se ha autorizado en pacientes con macroglobulinemia de Waldenström (MW) que han recibido al menos un tratamiento previo, o en primera línea para los pacientes que se consideren no adecuados para la inmuno-quimioterapia.
La indicación en MW en pacientes no candidatos a inmuno-quimioterapia en primera línea no se basa en datos directos, sino en la evidencia disponible en líneas posteriores de tratamiento.
La toxicidad farmacológica del ibrutinib es superponible a la experiencia más amplia encontrada en el tratamiento de los pacientes con LLC y LCM, sin haberse detectado en pacientes con MW nuevos efectos adversos. La linfocitosis y leucostasis vista en pacientes con LLC y LCM no ocurre en los pacientes con MW.
El perfil global de toxicidad del ibrutinib en la MW es favorable frente a la mayoría de opciones de monoterapia y de inmuno-quimioterapia, si bien se carece de estudios comparativos directos.
El ibrutinib en MW no se asocia a subidas (“flares”) de la IgM a diferencia de lo que ocurre con los tratamientos con rituximab.
INTRODUCCIÓN
La macroglobulinemia de Waldenström (MW) es una entidad clínico patológica definida poco frecuente que supone <2% de los linfomas no Hodgkin (1) con una incidencia de 0,4 casos por 100.000 habitantes y año (2, 3). Se clasifica dentro de los linfomas linfoplasmacíticos en la clasificación de la Organización Mundial de la Salud (OMS), aunque en otras clasificaciones esta entidad se incluye dentro de las gammapatías monoclonales. Para establecer el diagnóstico de MW se requiere demostrar una infiltración linfoplasmocítica en médula ósea y una IgM monoclonal (4).
La clínica de la MW incluye las manifestaciones relacionadas con la infiltración medular por el linfoma linfoplasmacítico, como citopenias (fundamentalmente anemia) y linfadenopatía /esplenomegalia, y las manifestaciones causadas por la paraproteína (síndrome de hiperviscosidad, crioglobulinemia, síndrome de aglutininas frías, polineuropatía, amiloidosis). La MW no tiene un pronóstico uniforme oscilando la supervivencia a 5 años desde un 85% a un 35%.
Las alteraciones moleculares de la MW son frecuentes y tienen importancia tanto en el diagnóstico como en el tratamiento. Las dos más frecuentes afectan a la señalización del receptor Toll-like y al CXCR4 (5). Molecularmente la MW se caracteriza por encontrase en el 90%-95% de los casos una única mutación (MYD88 L265P) tanto en casos sin tratamiento previo como aquellos tratados. El MYD88 es una molécula adaptadora que se encuentra en las vías de señalización del receptor Toll-like y del receptor de la interleucina 1 (6). La mutación MYD88 L265P desencadena la activación del NFkB a través de 2 vías que involucran a la tirosín quinasa de Bruton (BTK) y los receptores asociados a quinasas de la interleucina 1 (IRAKI 1 y IRAKI 4). En un tercio de los pacientes con MW se encuentran además de la mutación MYD88 L265P diversas mutaciones en el gen CXCR4 (se han descrito al menos 30) que induce la actividad de la BTK. Las mutaciones en CXCR4 encontradas en MW son similares a las que se encuentran en el síndrome WHIM (acrónimo inglés de: warts, hypogammaglobulinemia, infections, and myelokathexis).
En base a la combinación de estas mutaciones se han definido 3 grupos genómicos de MW: MYD88 L265P / CXCR4WT [WT indica tipo salvaje), MYD88 L265P /CXCR4WHIM, y MYD88WT /CXCR4) (7). El grupo genotípico de MW influye en la presentación clínica. Aquellos que no presentan mutación en MYD88 tienen peor supervivencia (7). Los pacientes con MYD88 no mutado no presentan generalmente mutaciones en CXCR4. Estas alteraciones moleculares han dado pie a nuevos tratamientos como son los inhibidores de la BTK (como el ibrutinib) o inhibidores del CRCX4 (como el plerixafor).
La MW se considera una enfermedad incurable, y dado que no hay evidencia del beneficio del tratamiento de pacientes asintomáticos con MW, el esperar y ver se considera el estándar actual para los casos asintomáticos (8).
- Se consideran indicaciones de tratamiento la presencia de síntomas causados por la IgM (como la hiperviscosidad, amiloidosis, crioglobulinemia, enfermedad por crioaglutininas, y neuropatía), la anemia (hemoglobina <10g/dl) o la trombopenia (<100.000 plaquetas/mm3) relacionada con la enfermedad, síntomas generales (fiebre, sudoración nocturna, pérdida de peso, fatiga), visceromegalia sintomática, y linfadenopatía sintomática o voluminosa (≥ 5 cm diámetro máximo) (9).
- El nivel de IgM per se no es criterio suficiente para iniciar el tratamiento (10).
- Las causas más frecuente de inicio de tratamiento en la MW son la anemia y la organomegalia.
En los pacientes con buen estado general el tratamiento recomendado tanto por el grupo de trabajo de la MW (9) como por las guías británicas (11) es la inmuno-quimioterapia, si bien existen diferencias en la recomendación de determinados regímenes terapéuticos.
Dado que el tratamiento actualmente disponible no es curativo tarde o temprano el paciente recidivará o progresará requiriendo un nuevo tratamiento. También hay pacientes refractarios al tratamiento inicial (12). Para estos dos supuestos no hay establecido un tratamiento estándar.
IBRUTINIB (IMBRUVICA®)
Ibrutinib (Imbruvica®) se encuentra autorizado para el tratamiento de los pacientes adultos con:
- Linfoma de células del manto (LCM) refractario o en recidiva.
- Leucemia linfocítica crónica (LLC) que hayan recibido al menos un tratamiento previo, o en primera línea para los casos que presenten del 17p o mutación TP53 en los que la inmuno- quimioterapia no se considere apropiada.
- MW en pacientes que han recibido al menos un tratamiento previo, o en primera línea para los pacientes que se consideren no adecuados para la inmuno-quimioterapia.
Esta última indicación es el objeto del presente informe.
La dosis aprobada de ibrutinib en MW es de 420 mg/día. El tratamiento se administra hasta progresión o hasta la aparición de toxicidad no tolerada.
Ibrutinib está designado como medicamento huérfano.
Farmacología
Ibrutinib, es un potente e irreversible inhibidor de la tirosín quinasa de Bruton (BTK).
Eficacia
Se dispone de dos estudios para el análisis de dosis-respuesta del fármaco: un estudio fase I (PCYC-04753) en pacientes con diversos síndromes linfoproliferativos que incluyó a 4 pacientes con MW, y un estudio fase Ib/II (PCYC-1102) (13). La dosis elegida para el estudio fase II en MW (14) se basó en los resultados del estudio PCY-1102 (13) donde se comprobó, en pacientes con LLC, que la saturación de la BTK se producía a dosis de 420 mg, sin observarse ventajas clínicas significativas al emplear dosis más elevadas (840 mg).
El registro para la MW se basa en el estudio PCYC-1118E, un estudio fase 2, de un solo brazo, realizado en 3 centros de EEUU, no comparativo en 63 pacientes con MW previamente tratados que requerían nuevo tratamiento (14).
Los pacientes en tratamiento anticoagulante con warfarina fueron excluidos, al igual que en el resto de estudios con ibrutinib en otras patologías. Los pacientes recibieron ibrutinib 420 mg/día en dosis única hasta progresión o el desarrollo de toxicidad inaceptable, o hasta un máximo de 40 ciclos (se consideró ciclo al tratamiento de 4 semanas). Los pacientes se siguieron hasta 2 años tras completar el estudio, hasta nuevo tratamiento o el fallecimiento, lo que ocurriera antes.
El objetivo primario fue la respuesta global evaluada por el investigador de acuerdo con los criterios del tercer taller internacional de MW (15), considerándose respuesta al menos una respuesta menor (lo que implica ≥ 25% reducción de los niveles de IgM).
Los objetivos secundarios fueron la obtención de respuestas mayores (RP o mejor, lo que implica ≥ 50% reducción de los niveles de IgM), duración de la respuesta, tiempo a respuesta, PFS, supervivencia global y mejoría en cifras de hemoglobina.
La edad mediana de los pacientes fue de 63 años (44-86), el 49% tenían ≥65 años. La mediana de tratamientos previos fue de 2 (1-11). Al momento de iniciar el tratamiento el 60% tenían anemia (≤11 g/dl) y el valor mediana de IgM sérica fue de 3,5 g/dl. La mediana de duración del tratamiento fue de 19,1 meses (0,5 – 29,7).
Los resultados del estudio fueron satisfactorios en cuanto a eficacia. La respuesta global fue del 90,5%, con un 73% de respuestas mayores: VGPR 10 casos (15,9%), PR 36 (57,1%) y MR 11 (17,5%), con una mediana de duración de la respuesta no alcanzada. No hubo casos de RC.
La mediana del tiempo a respuesta fue breve, 1 mes (0,7 a 13,4 meses). La mediana a respuesta mayor fue de 1,6 meses (0,72 – 13,4).
La respuesta obtenida fue similar analizando distintas características (edad, score pronóstico internacional, B2- microglobulina, cifra de hemoglobina, nivel de IgM, grado de infiltración medular, enfermedad refractaria o recidivante, y número de tratamientos previos). En cambio si que se observaron diferencias en el grado de respuesta de acuerdo a los 3 tipos genómicos de MW, clasificados en base al estado de mutación MYD88 y CXCR4 descritos(11). La respuesta fue mayor en los pacientes MYD88 L265P / CXCR4WT [WT indica tipo salvaje) (34 pacientes) (100% respuesta y 91% de respuestas mayores), seguido del grupo MYD88 L265P /CXCR4WHIM (85,7% respuestas y 61,9% de respuestas mayores) (21 pacientes) y por último el grupo MYD88WT /CXCR4 WT (71,4% globales; 28,6% mayores) (7 pacientes). La respuesta de IgM y hemoglobina fueron inicialmente discordantes con la respuesta en médula ósea, observándose descensos de IgM y ascensos de hemoglobina sin cambios en la carga tumoral medular.
La enfermedad extramedular también respondió de forma satisfactoria observándose una disminución de adenopatías en el 68% de los casos y de la esplenomegalia en el 57%. Los 4 casos tratados por presentar síndrome de hiperviscosidad respondieron al ibrutinib. Nueve pacientes tenían neuropatía progresiva relacionada con la IgM, observándose mejoría de la misma en 5 de ellos y estabilización en el resto.
La PFS y OS a 2 años fue del 69,1% y 95,2% respectivamente. No se observaron aumentos de IgM (“flares”) en estos casos de MW tratados con ibrutinib.
Seguridad
El perfil de seguridad del ibrutinib se basa en la experiencia del tratamiento en 420 pacientes: 246 con LLC, 111 con LCM y los 63 pacientes del estudio fase 2 en MW (14). En monoterapia y a largo plazo hay datos en 198 pacientes con solo 4 de ellos con MW.
De los 198 pacientes tratados en monoterapia con ibrutinib a largo plazo, con una mediana de tratamiento de 2 años, no se han detectado evidencia de toxicidad acumulativa.
Los efectos adversos más comunes (≥20%) encontrados en pacientes tratados con ibrutinib fueron: neutropenia, anemia, diarrea, dolor musculo-esquelético, infecciones del tracto respiratorio superior, sangrado, rash, náuseas y fiebre. Los efectos adversos grado 3 /4 más frecuentes (≥5%) fueron anemia, neutropenia, neumonía y trombopenia.
De los 420 pacientes tratados con ibrutinib el 59% tenían más de 65 años. Se observaron efectos adversos grado ≥3 con mayor frecuencia en pacientes >65años (53% vs 44% en más jóvenes).
La tasa de descontinuación de medicación por reacciones adversas fue del 12% en los 420 pacientes tratados con ibrutinib y el 9% en los pacientes con MW (14). Hubo un 9% de reacciones adversas que motivaron una reducción de dosis en la población general tratada y del 11% en la población con MW.
Infecciones grado 3-4 ocurrieron en el 14% de la población de 420 pacientes, y en el 9% del estudio en MW (14). La neutropenia grados 3-4 ocurrió en el 17% de la población de 420 pacientes, y en el 14% de los pacientes con MW (14).
Uno de los efectos adversos informados y sobre el que hay que observar una especial atención son las complicaciones hemorrágicas secundarias con y sin trombopenia. Se han descrito hemorragias gastrointestinales, intracraneales y hematuria, además de sangrados menores (petequias, epistaxis). Hemorragias grados 3-4 se observaron en el 3,6% de los 420 pacientes tratados, y en el 1,6% (1 caso en relación con biopsia de médula ósea) en el estudio de MW (14). Solo ha ocurrido un caso de hemorragia fatal (hematoma subdural en paciente trombopénico, 30 días tras la última dosis de ibrutinib suspendido por progresión de la MW). Si bien el mecanismo causante del sangrado por ibrutinib queda por aclarar de forma definitiva se sabe que el ibrutinib produce alteración de la función plaquetar (16). El ibrutinib afecta a la función plaquetar mediante la inhibición de la BTK y probablemente por la inhibición de la quinasa TEC (que también se ve inhibida irreversiblemente por el ibrutinib) (17). Por estudios in vitro, la transfusión de plaquetas frescas tras eliminar el ibrutinib puede corregir el defecto hemostático (16, 17). Los pacientes en tratamiento con anticoagulantes anti-vitamina K se excluyeron de los ensayos fase 2 y 3 con ibrutinib. No se deben administrar anticoagulantes anti- vitamina K en los pacientes que reciban ibrutinib. Debe evitarse el aceite de pescado y la vitamina E. El uso de otros anticoagulantes o fármacos que alteren la función plaquetar pueden aumentar el riesgo de sangrado de los pacientes tratados con ibrutinib por lo que se debe tener particular cuidado en estos casos si se emplean concomitantemente. No se han estudiado los efectos del ibrutinib en pacientes con diátesis hemorrágicas congénitas. Por el aumento de riesgo hemorrágico se recomienda suspender el ibrutinib de 3-7 días antes y después de la cirugía dependiendo del tipo de cirugía y el riesgo de hemorragia.
Recientemente se han revisado las complicaciones hemorrágicas del estudio RESONATE (18), en el que se comparaba ibrutinib vs ofatumumab en pacientes con LLC, y del estudio PCYC-1102 (13), un fase II en LLC. La incidencia de sangrado mayor (grado ≥3) global fue del 2,4% en los 327 pacientes tratados con ibrutinib (8 casos), similar al comparar ibrutinib con ofatumumab (un agente que no se asocia a diátesis hemorrágica): 2,4% con ibrutinib vs 1,6% con ofatumumab(19). De los 8 casos de sangrado con ibrutinib, 5 tenían tratamiento concomitante con anticoagulante (heparina bajo peso, 1 caso), antiagregante plaquetar (2 casos), o ambos (2 casos). El sangrado condujo a la discontinuación de la medicación en 4 pacientes (1,2%).
La linfocitosis, un fenómeno que ocurre frecuentemente al tratar pacientes con LLC (70%) o linfoma del manto (30%), no se ha observado en los pacientes tratados por MW. Se ha informado de unos pocos casos asociados a cifras extremadamente altas de linfocitos (> 400.000/mm3) en pacientes con LLC/LCM. No se han informado de casos de leucostasis en MW.
Se ha informado fibrilación auricular y fluter en el 4.5% de pacientes tratados con ibrutinib, especialmente en pacientes con factores de riesgo vascular. Dada la edad avanzada de los pacientes con MW es frecuente que presenten fibrilación auricular. Si ésta requiere anticoagulación se deben considerar otras opciones al ibrutinib. En el estudio de ibrutinib en MW (14) 3 casos desarrollaron fibrilación auricular, que tras un tiempo se resolvió pudiéndose continuar el tratamiento con ibrutinib en los 3 casos.
Como resumen del perfil de toxicidad del ibrutinib en MW, la toxicidad farmacológica del ibrutinib es superponible a la experiencia más amplia encontrada en el tratamiento de los pacientes con LLC y LCM, sin haberse detectado en pacientes con MW nuevos efectos adversos. La linfocitosis y leucostasis vista en pacientes con LLC y LCM no ocurre en los pacientes con MW. El ibrutinib en MW no se asocia a subidas (“flares”) de la IgM a diferencia de lo que ocurre con los tratamiento con rituximab.
DISCUSIÓN
En ficha técnica no hay fármacos que específicamente tengan a la MW como indicación. Sólo el clorambucilo tiene una indicación amplia en linfomas, y la bendamustina está indicada en linfomas indolentes no-Hodgkin que hayan progresado durante o en los 6 meses siguientes a un tratamiento con rituximab o un régimen que contenga rituximab. Por tanto el ibrutinib es actualmente el único fármaco con indicación específica en ficha técnica para la MW.
El tratamiento de pacientes en buen estado general con esta patología es la inmuno-quimioterapia, si bien existen diferencias en la recomendación de determinados regímenes terapéuticos. El R- CHOP no se considera ahora una opción de primera elección en la MW ya que ha resultado inferior comparado con regímenes con análogos de purina en monoterapia (20) o bendamustina (21), y por la frecuente neurotoxicidad de la vincristina (9). Además el uso de regímenes sin adriamicina no parecen disminuir las respuestas o su duración (12). La neuropatía es uno de los efectos preocupantes del bortezomib en la MW. En pacientes con MW el tratamiento en primera línea con bortezomib bisemanal se asocia a neuropatía ≥ grado 2 en el 70% de los casos, teniendo que discontinuar el tratamiento en el 61% de los pacientes (22). Su uso semanal y por vía subcutánea reduce la aparición de neuropatías. Su uso semanal se asocia a una neuropatía de ≥ grado 2 en el 24%, discontinuando el tratamiento por este motivo solo el 8% de los casos (23). La seguridad a largo plazo de las combinaciones con bortezomib en MW no se conocen.
Los tratamiento de inmuno-quimioterapia recomendados por el grupo de trabajo de la MW (9) son la combinación de rituximab con ciclofosfamida y dexametasona (DRC), rituximab con bortezomib y dexametasona (BDR), y rituximab con bendamustina (BR). El grupo de trabajo de la MW no recomienda para el tratamiento de primera línea las combinaciones con fludarabina pese a sus buenas respuestas (similar al resto de regímenes de combinación con rituximab) aunque son una opción para los casos recidivantes o refractarios. La mielotoxicidad, la posible interferencia en recolección de progenitores para un auto-trasplante y los efectos a largo plazo son razones que limitan su uso. Los regímenes DRC y BR son menos tóxicos que el FCR e igualmente efectivos. El uso de análogos nucleósidos en la MW se ha asociado con un aparente mayor riesgo de transformación a linfoma agresivo o a SMD/LAM (24). El régimen FCR (fludarabina /ciclofosfamida /rituximab) se asocia con mielosupresión prolongada cuando se administran 6 o más ciclos, por lo que no se recomienda emplear más de 5 ciclos.
En cambio en las guías británicas si se indican en primera línea análogos nucleósidos: FCR (fludarabina + ciclofosfamida + rituximab) y cladribina + rituximab (11). Las guías británicas no recomiendan bortezomib en el tratamiento inicial (a diferencia de la recomendación del grupo de trabajo de MW), aunque sí para casos en recidiva (11).
En los pacientes que no pueden tolerar combinaciones de rituximab, las opciones en uso son fludarabina, rituximab y clorambucilo.
En las guías internacionales de MW se prefiere la fludarabina en monoterapia sobre el clorambucilo (9). Esta recomendación se basa en un estudio randomizado que mostró que la fludarabina en monoterapia era superior al clorambucilo asociándose con tendencia a más respuestas (46% vs 36%, P 0,07), mayor duración de las mismas (38 vs 21 meses, P 0,002), incremento en supervivencia global a 5 años (69% vs 62%, P 0,046) y menor desarrollo de segundos tumores (4% vs 21%, P 0,001) (25).
Otra opción en monoterapia es el rituximab que obtiene unas respuestas entre el 25-45% (8). Hay que tener en cuenta que el rituximab en monoterapia se asocia en un 40-50% con un incremento inicial del nivel de la IgM lo que puede ser perjudicial (8). Su asociación con quimioterapia o bortezomib, pero no con talidomida, evita o disminuye este incremento. Una IgM >4 g/dl o una viscosidad de > 4 centipoises se consideran situaciones de riesgo de desarrollar síndrome de hiperviscosidad, y se recomienda diferir el inicio de rituximab en estos casos dando previamente otros tratamientos (plasmaféresis, bortezomib, etc.).
Para el tratamiento de la neuropatía asociada a IgM el rituximab en monoterapia no mostró ser más eficaz que el placebo (26).
En el caso de pacientes refractarios al tratamiento inicial, las opciones de tratamiento pueden ser diversas:
En general se considera que los regímenes de rituximab con quimioterapia son los tratamientos a emplear.
Puede repetirse el régimen empleado en el anterior tratamiento si la respuesta fue satisfactoria y la recidiva/progresión no ocurre precozmente (≥12 meses de respuesta para el grupo de trabajo de MW) (9) (o ≥24 meses en las guías británicas) (11).
Si la recaída ocurre precozmente o el paciente es refractario se debe emplear un régimen terapéutico distinto al inicial. Si el anterior ciclo fue de rituximab más alquilante se recomienda emplear otro con rituximab con análogo nucleósido, rituximab con bortezomib o rituximab con bendamustina.
Si se considera el trasplante autólogo una opción para el paciente hay que evitar el uso repetido de análogos nucleósidos.
El trasplante autólogo es una opción para los pacientes que recidivan en los 6 primeros meses tras rituximab más quimioterapia, si el paciente es quimiosensible y tiene una edad adecuada < 65-70 años. El trasplante autólogo consigue una PFS y OS a 5 años del 40% y 68% respectivamente y una tasa de RC/VGPR del 70% (27). A diferencia de lo que ocurre en el mieloma múltiple, el trasplante autólogo se infrautiliza en la MW pese a las buenas respuestas obtenidas y a la experiencia positiva acumulada (28). El TPH autólogo está indicado en pacientes de edad adecuada con MW en recidiva con enfermedad quimiosensible o en primera línea en pacientes de muy alto riesgo (9).
El trasplante alogénico se considera investigacional en la MW, siendo escasa la experiencia publicada (8).
La valoración de la respuesta al tratamiento se hace con unos criterios específicos de consenso internacional para la MW (29). De acuerdo a estos criterios existen 6 categorías de respuesta (remisión completa -CR-, muy buena remisión parcial -VGPR-, remisión parcial –PR-, respuesta menor –MR-, enfermedad estable –SD-, y progresión –PD-). La remisión completa requiere, entre otros criterios, la ausencia de paraproteína IgM detectable por inmunofijación. Se considera que todos los ensayos deben dar al menos información sobre supervivencia global, supervivencia libre de progresión (PFS) y tiempo a progresión (TTP), y en aquellos pacientes con respuesta el tiempo de duración de la respuesta (DOR), y en los que obtienen RC la supervivencia libre de enfermedad (DFS). Se recomienda igualmente informar sobre el tiempo hasta el siguiente tratamiento (TTNT) así como la supervivencia causa-específica (CSS). La cinética de respuesta de la IgM varía dependiendo del tipo de tratamiento aplicado, por lo que hay que tenerlo en cuenta a la hora de valorar la respuesta (11, 9). Los anticuerpos monoclonales y los análogos de las purinas producen un descenso lento de la IgM con depleción de la infiltración medular por células CD20+, mientras que el bortezomib produce el efecto opuesto. Para poner en perspectiva los resultados del ibrutinib en MW (14) solo se puede hacer comparación histórica con resultados de otros estudios ya que a fecha de hoy no hay estudios comparativos con otros fármacos en esta patología.
En monoterapia el ibrutinib obtiene mejores respuestas que la mayoría de otros tratamientos en monoterapia o similares con un perfil de toxicidad superior (tabla 1).
El ibrutinib obtiene mayores tasas de respuestas que las publicadas para las opciones más empleadas en monoterapia (tabla 1) como son el clorambucilo (25), fludarabina (25), cladribina (30), rituximab (31), alemtuzumab (32), y similares o superiores a las obtenidas con everolimus (33) y bortezomib (34).
En relación al estado mutacional, los resultados obtenidos con ibrutinib parecen indicar que el estado de la mutación en MYD88 marca el grado y profundidad de respuesta, siendo mayores en los casos mutados (MYD88 L265P). La mutación en CXCR4 disminuye la respuesta y por tanto podría ser un escenario en el que valorar otras alternativas de tratamiento. Sin embargo, en ese caso, debe tenerse en cuenta que:
- en los estudios con ibrutinib, la respuesta en la mutación CXCR4 se mantiene en porcentajes elevados a pesar del patrón genético de mal pronóstico, si bien el número de pacientes de los diferentes subgrupos de patrón de mutaciones incluidos en los estudios es limitado.
- la posible menor respuesta podría hacerse extensible a otras alternativas de tratamiento (35).
Tabla 1: Estudios de monoterapia en MW
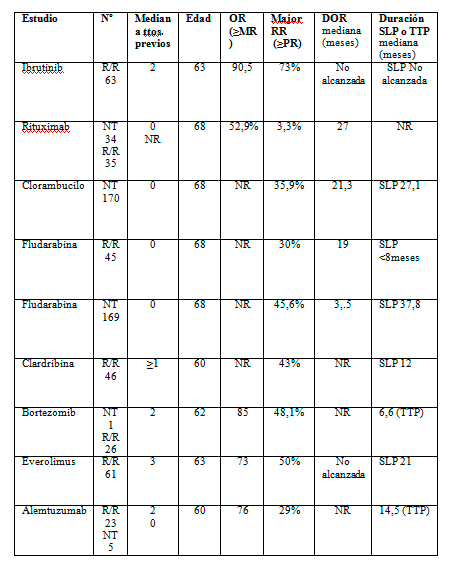
SLP: supervivencia libre de progresión; TTP: tiempo a progresión; NR: no informado; NT: no tratados previamente
Con everolimus (33) se observó significativa toxicidad, con un 67% de los pacientes con toxicidad grado ≥ 3 relacionados con el fármaco. Un 5% desarrollaron neumonitis que motivó discontinuación o suspensión del fármaco. El 17% presentó neutropenia grado 3-4 y un 20% trombopenia grado 3-4. El 62% de los pacientes tuvieron que reducir dosis, y el 40% tuvo que retrasar el tratamiento debido fundamentalmente a citopenias.
En el estudio con alemtuzumab (32) la toxicidad fue significativa: el 48% de los pacientes abandonaron el tratamiento por diversas toxicidades. En los pacientes que habían recibido tratamientos previos, el 74% desarrollaron toxicidad grado ≥3. El 54% de los pacientes tuvieron neutropenia grado ≥ 3, y el 25% trombopenia grado ≥3. El 18% desarrolló viremia por CMV. Tres pacientes (11%) fallecieron por causas infecciosas.
La PFS parece más prolongada con el ibrutinib que en el resto de opciones en monoterapia (figura 7). Hay que tener en cuenta que gran parte de estos estudios en monoterapia se han realizado en pacientes sin tratamiento previo, a diferencia del ibrutinib, o son una mezcla de pacientes no tratados previamente con casos recidivantes/refractarios, lo que dificulta la comparación.
Figura 7: Comparación histórica de SLP en pacientes con MW (uso de un único medicamento)

La mayor parte de estudios de combinación en MW se han realizado en pacientes sin tratamiento previo. A fecha de hoy no existen tratamientos combinados publicados con ibrutinib en MW si bien está en curso un estudio fase III (PCYC-1127) randomizado de rituximab con y sin ibrutinib (clinicaltrials identificador: NCT02165397).
En pacientes refractarios/recidivantes la combinación de bendamustina más rituximab (36) consiguió un 83% de respuestas con un tiempo mediano a progresión de 13,2 meses (tabla 2).
Con el régimen FCR en 43 pacientes (37), la mayoría tratamientos iniciales (65%) se obtuvieron un 79% respuestas globales con un 74% de mayores, con una mediana de SLP de 50 meses. Los pacientes con recidiva/refractarios obtuvieron respuestas peores aunque no están analizados de forma separadas para los casos tratados en primera línea vs R/R las respuestas globales, respuesta mayores ni SLP. La toxicidad fue considerable presentando neutropenia 3-4 en el 88% de los pacientes pese a emplear G-CSF en el 65% de los ciclos, el 44% presentó neutropenia prolongada (mediana 7 meses), el 35% de los pacientes recibieron menos de los 6 ciclos programados por diversas toxicidades, el 14% (6 pacientes) tuvieron infecciones graves, 2 pacientes fallecieron por infección.
El régimen de rituximab con fludarabina (38) da unos resultados similares al FCR (95% respuestas, 86% respuestas mayores) pero se asocia a toxicidad considerable: neutropenia grados 3-4 en el 63%, infecciones grados 3-4 en el 16% con 2 muertes por P.jiroveci, y desarrollo de SMD/LAM/otros tumores en el 18% de los pacientes.
El régimen DRC obtiene en primera línea buenos resultados (39) con escasa toxicidad (neutropenia grados 3-4 del 9%) (tabla 2).
El carfilzomib, un inhibidor de proteosoma de segunda generación con clara menor producción de neuropatía que el bortezomib (40) se estudió recientemente en la MW como tratamiento de primera línea asociado a rituximab y dexametasona (41). Se obtuvo un 87% de respuestas globales con un 68% de respuestas mayores. Lo más interesante fue la baja incidencia de neuropatía (solo 1 caso de grado 2, ninguno de grado 3-4).
Se puede concluir que el perfil global de toxicidad del ibrutinib en la MW es favorable frente a la mayoría de opciones de monoterapia y de inmuno-quimioterapia, si bien se carece de estudios comparativos directos.
Tabla 2: Estudios de terapia combinada en MW
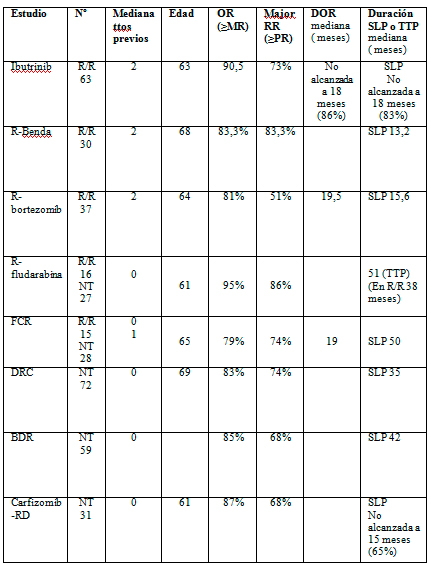
SLP: supervivencia libre de progresión; TTP: tiempo a progresión; NR: no informado; NT: no tratados previamente; FCR: fludarabina, ciclofosfamida, rituximab; DRC: dexametasona, rituximab, ciclofosfamida.
CONCLUSIÓN
Ibrutinib es el primer medicamento con indicación específica para la MW en pacientes adultos.
En pacientes con MW recidivante o refractaria, el ibrutinib en monoterapia dada la combinación de eficacia y perfil de toxicidad, es una nueva opción a emplear en estos pacientes, debiendo adecuarse su uso a las características del pacientes (empleo de anticoagulantes y antiagregantes, fundamentalmente).
La extensión de la indicación a MW en primera línea, para los pacientes que se consideren no adecuados para la inmuno- quimioterapia, no se basa en datos directos proporcionados por el estudio de registro (realizado en pacientes en recidiva o refractarios) sino en el razonamiento de que no hay base para esperar menor eficacia o pero perfil de seguridad en primera línea que en los casos recidivantes o refractarios.
CONSIDERACIONES FINALES DEL GCPT
Tras la decisión de financiación y precio, no se han identificado aspectos que puedan modificar el posicionamiento de Imbruvica.
REFERENCIAS
1. Wang H, Chen Y, Li F, Delasalle K, Wang J, Alexanian R, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: a large population-based study. Cancer. 2012; 118(15):3793-800.
2. The Surveillance E, and End Results (SEER) Program of the National Cancer Institute. All Lymphoid Neoplasms With Detailed Non-Hodgkin Lymphoma Subtypes 2015 [11 may 2015]. Available from: http://seer.cancer.gov/archive/csr/1975_2010/results_merged/sect_19_nhl.pdf.
3. Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006; 107(1):265-76.
4. Owen RG, Treon SP, Al-Katib A, Fonseca R, Greipp PR, McMaster ML, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30(2):110-5
5. Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637-46.
6. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012; 367(9):826-33.
7. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014; 123(18):2791-6.
8. National Comprehensive Cancer Network. Waldesntröm's Macroglobulinemia Lymphoplasmatic lymphoma. NCCN V.3.2015. NCCN Clinical Practice Guidelines in Oncology [Internet]. 2015. Available from: http://www.nccn.org/professionals/physician_gls/pdf/waldenstroms.pdf.
9. Dimopoulos MA, Kastritis E, Owen RG, Kyle RA, Landgren O, Morra E, et al. Treatment recommendations for patients with Waldenstrom macroglobulinemia (WM) and related disorders: IWWM-7 consensus. Blood. 2014; 124(9):1404-11.
10. D'Souza A, Ansell S, Reeder C, Gertz MA. Waldenstrom macroglobulinaemia: the key questions. Br J Haematol. 2013; 162(3):295-303.
11. Owen RG, Pratt G, Auer RL, Flatley R, Kyriakou C, Lunn MP, et al. Guidelines on the diagnosis and management of Waldenström macroglobulinaemia. British Journal of Haematology. 2014; 165(3):316-33.
12. Buske C, Leblond V. How to manage Waldenstrom's macroglobulinemia. Leukemia. 2013;27(4):762-72.
13. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013; 369(1):32-42.
14. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. New England Journal of Medicine. 2015; 372(15):1430-40.
15. Kimby E, Treon SP, Anagnostopoulos A, Dimopoulos M, Garcia-Sanz R, Gertz MA, et al. Update on recommendations for assessing response from the Third International Workshop on Waldenstrom's Macroglobulinemia. Clin Lymphoma Myeloma. 2006; 6(5):380-3.
16. Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015; 29(4):783-7.
17. Levade M, David E, Garcia C, Laurent PA, Cadot S, Michallet AS, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014; 124(26):3991-5.
18. Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. New England Journal of Medicine. 2014; 371(3):213-23.
19. Jones JA, Hillmen P, Coutre S, Tam C, Furman RR, Barr PM, et al. Pattern of Use of Anticoagulation and/or Antiplatelet Agents in Patients with Chronic Lymphocytic Leukemia (CLL) Treated with Single-Agent Ibrutinib Therapy. Blood. 2014; 124(21):1990-.
20. Leblond V, Levy V, Maloisel F, Cazin B, Fermand JP, Harousseau JL, et al. Multicenter, randomized comparative trial of fludarabine and the combination of cyclophosphamide- doxorubicin-prednisone in 92 patients with Waldenstrom macroglobulinemia in first relapse or with primary refractory disease. Blood. 2001; 98(9):2640-4.
21. Rummel MJ, Niederle N, Maschmeyer G, Banat GA, von Grunhagen U, Losem C, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013.
22. Treon SP, Ioakimidis L, Soumerai JD, Patterson CJ, Sheehy P, Nelson M, et al. Primary therapy of Waldenstrom macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol. 2009; 27(23):3830-5.
23. Dimopoulos MA, Garcia-Sanz R, Gavriatopoulou M, Morel P, Kyrtsonis MC, Michalis E, et al. Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood. 2013; 122(19):3276-82.
24. 24. Leleu X, Soumerai J, Roccaro A, Hatjiharissi E, Hunter ZR, Manning R, et al. Increased Incidence of Transformation and Myelodysplasia/Acute Leukemia in Patients With Waldenström Macroglobulinemia Treated With Nucleoside Analogs. Journal of Clinical Oncology. 2009; 27(2):250-5.
25. Leblond V, Johnson S, Chevret S, Copplestone A, Rule S, Tournilhac O, et al. Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenstrom macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol. 2013; 31(3):301-7.
26. Leger JM, Viala K, Nicolas G, Creange A, Vallat JM, Pouget J, et al. Placebo-controlled trial of rituximab in IgM anti-myelin- associated glycoprotein neuropathy. Neurology. 2013; 80(24):2217-25.
27. Kyriakou C, Canals C, Sibon D, Cahn JY, Kazmi M, Arcese W, et al. High-Dose Therapy and Autologous Stem-Cell Transplantation in Waldenstrom Macroglobulinemia: The Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010; 28(13):2227-32.
28. Gertz MA, Reeder CB, Kyle RA, Ansell SM. Stem cell transplant for Waldenstrom macroglobulinemia: an underutilized technique. Bone Marrow Transplant. 2012; 47(9):1147-53.
29. Owen RG, Kyle RA, Stone MJ, Rawstron AC, Leblond V, Merlini G, et al. Response assessment in Waldenstrom macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013; 160(2):171-6.
30. Dimopoulos MA, Weber D, Delasalle KB, Keating M, Alexanian R. Treatment of Waldenstrom's macroglobulinemia resistant to standard therapy with 2-chlorodeoxyadenosine: identification of prognostic factors. Ann Oncol. 1995; 6(1):49- 52.
31. Gertz MA, Rue M, Blood E, Kaminer LS, Vesole DH, Greipp PR. Multicenter phase 2 trial of rituximab for Waldenstrom macroglobulinemia (WM): an Eastern Cooperative Oncology Group Study (E3A98). Leuk Lymphoma. 2004; 45(10):2047-55.
32. Treon SP, Soumerai JD, Hunter ZR, Patterson CJ, Ioakimidis L, Kahl B, et al. Long-term follow-up of symptomatic patients with lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia treated with the anti-CD52 monoclonal antibody alemtuzumab. Blood. 2011; 118(2):276-81.
33. Ghobrial IM, Witzig TE, Gertz M, LaPlant B, Hayman S, Camoriano J, et al. Long-term results of the phase II trial of the oral mTOR inhibitor everolimus (RAD001) in relapsed or refractory Waldenstrom Macroglobulinemia. Am J Hematol. 2014; 89(3):237-42.
34. Treon SP, Hunter ZR, Matous J, Joyce RM, Mannion B, Advani R, et al. Multicenter clinical trial of bortezomib in relapsed/refractory Waldenstrom's macroglobulinemia: results of WMCTG Trial 03-248. Clin Cancer Res. 2007; 13(11):3320-5.
35. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's Macroglobulinemia. Leukemia. 2015; 29(1):169-76.
36. Treon SP, Hanzis C, Tripsas C, Ioakimidis L, Patterson CJ, Manning RJ, et al. Bendamustine therapy in patients with relapsed or refractory Waldenstrom's macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011; 11(1):133-5.
37. Tedeschi A, Benevolo G, Varettoni M, Battista ML, Zinzani PL, Visco C, et al. Fludarabine plus cyclophosphamide and rituximab in Waldenstrom macroglobulinemia: an effective but myelosuppressive regimen to be offered to patients with advanced disease. Cancer. 2012; 118(2):434-43.
38. Treon SP, Branagan AR, Ioakimidis L, Soumerai JD, Patterson CJ, Turnbull B, et al. Long-term outcomes to fludarabine and rituximab in Waldenstrom macroglobulinemia. Blood. 2009; 113(16):3673-8.
39. Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, Zervas K, Tsatalas C, Kokkinis G, et al. Primary treatment of Waldenstrom macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol. 2007; 25(22):3344-9.
40. Siegel D, Martin T, Nooka A, Harvey RD, Vij R, Niesvizky R, et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase 2 clinical studies. Haematologica. 2013; 98(11):1753-61.
41. Treon SP, Tripsas CK, Meid K, Kanan S, Sheehy P, Chuma S, et al. Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenstrom's macroglobulinemia. Blood. 2014; 124(4):503-10.
GRUPO DE EXPERTOS
(por orden alfabético)
Agencia Española de Medicamentos y Productos Sanitarios
Rafael de la Cámara
Servicio de Hematología. Hospital de la Princesa, Madrid.
Subdirección General de Posicionamiento Terapéutico y Farmacoeconomía
Dirección General de Farmacia y Productos Sanitarios. Conselleria de Sanitat. Valencia.
Todos los expertos han realizado una declaración de conflictos de interés.
El Laboratorio Titular, la Sociedad Española de Hematología y Hemoterapia, Sociedad Española de Farmacología Clínica, Sociedad Española de Farmacia Hospitalaria, la Asociación Linfoma Mieloma y Leucemia han tenido oportunidad de enviar comentarios al documento, si bien el texto final es el adoptado por el GCPT.
Fuente: Agencia Española de Medicamentos y Productos Sanitarios.
