Criterios y recomendaciones generales para el tratamiento con boceprevir y telaprevir de la hepatitis crónica C en pacientes co-infectados por el VIH y trasplantados hepáticos
INFORME DE POSICIONAMIENTO TERAPÉUTICO PT-BOC/TEL-COINF-TRAS/V3/01112014
Criterios y recomendaciones generales para el tratamiento con boceprevir y telaprevir de la hepatitis crónica C en pacientes co- infectados por el VIH y trasplantados hepáticos
Fecha de publicación: 18 de noviembre de 2014
La infección por el virus de la hepatitis C (VHC) es un grave problema de salud en el mundo occidental donde la prevalencia de infección crónica por VHC varía entre el 1,5 y el 3% de la población. El tratamiento de la hepatitis crónica C (HCC), con la combinación de interferón pegilado (P-INF) alfa 2a ó 2b y ribavirina (RBV), consigue una respuesta viral sostenida (RVS) en aproximadamente el 50 % de pacientes infectados por el genotipo 1 del VHC que es el más común en nuestro entorno (75% de los infectados).
Boceprevir y telaprevir, los primeros agentes anti-VHC directos comercializados, están indicados, en combinación con P-INF y RBV, para el tratamiento de pacientes adultos con HCC (genotipo 1) con enfermedad hepática compensada.
En pacientes mono-infectados, la triple terapia basada en P-INF alfa, RBV y un agente antiviral directo como boceprevir o telaprevir mejora la tasa de RVS hasta un 75 % (pacientes naïve) y hasta un 50 % en el caso de pacientes que han fracasado al tratamiento previo. Sin embargo, la triple terapia puede asociarse a ciertas reacciones adversas tales como exantema cutáneo (telaprevir) que puede ser grave hasta en un 5% de los casos, anemia (telaprevir y boceprevir) o disgeusia (boceprevir). Por otro lado, la ausencia de respuesta a la triple terapia se asocia de manera casi universal al desarrollo de mutaciones de resistencia en el VHC. Estas mutaciones tienden a desaparecer de manera progresiva con el tiempo, pero su significado clínico es incierto ya que no hay ningún estudio que haya valorado la respuesta al tratamiento con otros antivirales de la misma familia de pacientes con variantes víricas con mutaciones de resistencia. Así mismo, se debe tener en cuenta que existen un número elevado de agentes antivirales directos en distintas fases de desarrollo (incluidos dos actualmente en fase de registro), que pueden estar disponibles en un futuro próximo y que pueden significar un incremento aún mayor de la tasa de respuesta, con menor tiempo de tratamiento y un menor número de efectos adversos. Por tanto, se debe adoptar una actitud prudente en cuanto a la incorporación a la práctica de estas primeras pautas triples.
En definitiva, la triple terapia tiene una eficacia superior a la del tratamiento clásico, pero ocasiona generalmente más efectos adversos e implica, además, la necesidad de una evaluación más minuciosa y un seguimiento más frecuente de los enfermos en tratamiento. Ambos hechos pueden tener un impacto asistencial importante en los centros que atienden enfermos con HCC así como en el coste asociado al tratamiento.
No hay estudios que hayan comparado directamente boceprevir y telaprevir por lo que la comparación entre ambos sólo puede ser indirecta. Con los datos actuales, el perfil beneficio/riesgo de ambos es similar cuando se usan ateniéndose a las recomendaciones que se establecen en este documento.
BOCEPREVIR Y TELAPREVIR EN PACIENTES CO-INFECTADOS POR EL VIH, TRASPLANTADOS DE HÍGADO Y NIÑOS/ADOLESCENTES
Boceprevir y telaprevir han sido autorizados por la Comisión Europea mediante un procedimiento centralizado en base a los resultados de ensayos clínicos en pacientes mono-infectados. El nivel de evidencia en pacientes co-infectados por el VIH y en trasplantados de hígado, ha aumentado de forma sustancial desde su autorización. Aún así, se espera recabar más información de los ensayos clínicos en marcha en estas poblaciones. Puede consultarse un listado completo y actualizado de los ensayos clínicos autorizados en España para el tratamiento de la hepatitis C en pacientes coinfectados por VIH y en pacientes con trasplante hepático en la dirección http://www.aemps.gob.es
De momento no se recomienda el uso de estos medicamentos en niños y adolescentes debido a la falta de datos de eficacia y seguridad.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha decidido elaborar las siguientes recomendaciones de uso para pacientes coinfectados y trasplantados. Dado el aumento del conocimiento generado en estas situaciones y la existencia de un sistema activo de farmacovigilancia, el registro obligatorio de casos de la AEMPS no seguirá operativo a partir de la fecha de publicación de este documento.
Los criterios se han elaborado partiendo de los utilizados para el programa de acceso precoz de la AEMPS que fueron elaborados por un grupo de expertos entre los que estaban los implicados en la evaluación de estos medicamentos para su autorización por parte de la AEMPS y clínicos expertos en el manejo de la HCC en pacientes monoinfectados, co-infectados por VIH y trasplantados hepáticos. A ellos se han sumado expertos designados por las Comunidades Autónomas de Cataluña, Madrid, Andalucía y Galicia (ver anexo el listado de autores al final del documento).
1. CRITERIOS DE USO PARA PACIENTES CO- INFECTADOS POR EL VIH Y VHC
Los pacientes co-infectados candidatos a triple terapia con P- INF, RBV y un inhibidor de la proteasa del VHC (boceprevir o telaprevir) han de cumplir todos los criterios siguientes (1 a 8)
Criterios dependientes del VHC
(1) Infección por VHC genotipo 1, independientemente de que el paciente haya recibido o no tratamiento previo para la HCC
(2) Fibrosis F2, F3 y F4 confirmada por biopsia hepática o rigidez hepática medida por Fibroscan ≥7.6 Kilopascales
(3) Determinación del polimorfismo IL28B únicamente en pacientes con fibrosis F2 antes del inicio del tratamiento.
En el caso particular de pacientes F2 con genotipo favorable CC, se recomienda iniciar tratamiento con P-INF y RBV, valorando la Respuesta Viral Rápida (RVR) definida como la ausencia de detección del ARN del VHC a las 4 semanas de tratamiento. Si hay RVR, se recomienda mantener el tratamiento con interferón pegilado y ribavirina durante 48 semanas. Si no hubiera RVR, se recomienda utilizar triple terapia.
En pacientes F3 y F4, independientemente del genotipo IL28B se recomienda iniciar tratamiento con triple terapia.
La información disponible acerca de la historia natural de la hepatitis C en pacientes infectados por VIH teniendo en cuenta el estadio de fibrosis hepática confirmado por biopsia indica que las tasas de eventos hepáticos (muerte hepática, descompensación y carcinoma hepatocelular) aumentan de forma gradual según los estadios de METAVIR. Así, el riesgo de padecer algún evento hepático a cinco años es bajo en pacientes con F0 o F1 y muy elevado para los pacientes con F4. Es destacable, sin embargo, que el riesgo de padecer algún evento hepático con ese horizonte temporal no es significativamente diferente para los pacientes con fibrosis F2 en comparación con los pacientes con fibrosis F3.
Antes de iniciar tratamiento con triple terapia en pacientes coinfectados por VIH/VHC con cirrosis hepática, debe tenerse en cuenta que en pacientes mono-infectados con cirrosis y enfermedad hepática avanzada –caracterizada por una cifra de plaquetas <100.000/mm3 y una albúmina sérica <3,5 g/dl– el riesgo de padecer efectos adversos graves (algunos de ellos mortales) aumenta significativamente. Aunque en el paciente co-infectado las alteraciones analíticas arriba mencionadas (en particular la trombocitopenia) no tienen el mismo significado clínico que en el mono-infectado, y en espera de más información al respecto, se recomienda valorar la existencia de alternativas terapéuticas más adecuadas para estos pacientes. En caso de iniciar el tratamiento con triple terapia, éste deberá llevarse a cabo en centros con experiencia en el manejo de cirróticos descompensados y con fácil accesibilidad al trasplante hepático.
(4) Hepatopatía compensada (Child-Pugh grado A)
(5) Concentración de hemoglobina >11 g/dl en mujeres y >12 g/dl en hombres
(6) Independientemente del estadio de fibrosis, serán candidatos a iniciar terapia triple aquellos pacientes coinfectados con manifestaciones extra-hepáticas graves de la infección por VHC como por ejemplo aquellas derivadas de la crioglobulinemia mixta.
Criterios dependientes del VIH
(7) Pacientes con tratamiento antirretroviral
- Linfocitos CD4+ totales en sangre periférica >100 /ml o porcentaje de linfocitos CD4+ >12%
- Carga viral plasmática de VIH <1000 copias/ml
(8) No se aplican los criterios anteriormente mencionados de CD4+ ni de carga viral plasmática del VIH en aquellos pacientes que no requieran tratamiento antirretroviral según las recomendaciones actuales de tratamiento del VIH.
Tratamiento antirretroviral concomitante1
Antirretrovirales permitidos con boceprevir
| Abacavir; Emtricitabina; Lamivudina; Tenofovir |
| Etravirina (200mg cada 12 horas)2,3 Rilpivirina (25mg una vez al día) |
| Atazanavir/ritonavir5,6 |
| Raltegravir |
| Maraviroc |
| No están permitidos por el momento |
Antirretrovirales permitidos con telaprevir
| Abacavir; Emtricitabina; Lamivudina; Tenofovir |
| Efavirenz (incrementando la dosis de telaprevir a 1.125 mg/8h) Etravirina3 Rilpivirina |
| Atazanavir/ritonavir |
| Raltegravir |
| Maraviroc (150mg/24h)7 |
| No están permitidos por el momento |
1 La información sobre el tratamiento antirretroviral permitido y no permitido se mantendrá actualizada en base a los datos procedentes de los diferentes ensayos clínicos actualmente en marcha.
2 Los parámetros farmacocinéticos de etravirina disminuyen, como también disminuye la Cmin de boceprevir. Se desconoce la relevancia clínica de estos hallazgos, por lo que está justificado un incremento de la vigilancia clínica y de los valores de laboratorio.
3 Ver sección Recomendaciones para el manejo del exantema.
4 No se recomienda el uso de darunavir, lopinavir/ritonavir y fosamprenavir
5 Únicamente en pacientes con carga viral del VIH suprimida y cepa viral del VIH sin sospecha de resistencia al tratamiento antirretroviral. Está justificado un incremento de la vigilancia clínica de la respuesta al tratamiento y de parámetros de laboratorio.
6 Se recomienda un seguimiento clínico y analítico por si aparece hiperbilirrubinemia
7 Los datos disponibles indican que el AUC de maraviroc aumenta con la administración de telaprevir y boceprevir (hasta 9 veces mayor con telaprevir y 3 veces mayor con boceprevir), mientras que la exposición a éstos no se modifica. Boceprevir y maraviroc pueden utilizarse conjuntamente a las dosis habituales. En caso de considerar el uso de telaprevir, se recomienda reducir la dosis de maraviroc a 150mg una vez al día, con el objetivo de reducir el riesgo de posibles eventos adversos, fundamentalmente prolongación del QT e hipotensión ortostática. Estas recomendaciones están avaladas por el grupo de expertos en base a los datos existentes y podrían ser revisadas en el futuro tras su evaluación formal por el Comité de Medicamentos de Uso Humano (CHMP).
NOTA: la ficha técnica de los medicamentos puede haber variado desde la publicación de este documento. La ficha técnica actualizada que contiene los últimos cambios incorporados se encuentra disponible en http://www.aemps.gob.es/
DOSIS Y PAUTAS DE ADMINISTRACIÓN EN PACIENTES COINFECTADOS POR VIH Y VHC
El tratamiento con ambos medicamentos debe ser iniciado y supervisado por un médico con experiencia en el manejo de la infección por VIH y de la hepatitis crónica C.
Boceprevir
La dosis habitual de boceprevir es de 800mg (4 cápsulas) cada 8 horas (se recomienda un intervalo de 7 a 9 horas), administrados con comida. Esta dosis debe modificarse como se ha indicado anteriormente dependiendo del tratamiento antirretroviral que esté recibiendo el paciente. Las cápsulas deben tragarse enteras.
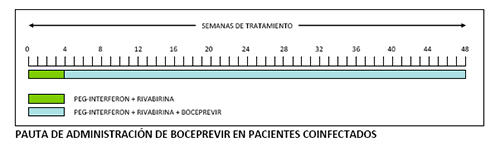
La duración total del tratamiento es de 48 semanas. Boceprevir debe administrarse en combinación con P-IFN alfa (2a o 2b) y RBV, iniciándose después de un periodo inicial de 4 semanas («lead-in») sólo con P-IFN y RBV. La triple terapia (boceprevir, P-IFN y RBV) debe mantenerse durante 44 semanas más hasta completar las 48 semanas totales de tratamiento.
A diferencia de lo que ocurre con telaprevir, la mayoría de los estudios de boceprevir se han realizado en combinación con P-INF alfa 2b.
Telaprevir
La dosis habitual de telaprevir es de 750mg (2 comprimidos) cada 8 horas, administrados con comida. Esta dosis debe modificarse como se ha indicado anteriormente dependiendo del tratamiento antirretroviral que esté recibiendo el paciente. Los comprimidos deben tragarse enteros (sin masticar o triturar o disolver). Aunque no existen datos en pacientes coinfectados y trasplantados, telaprevir se podrá administrar también a dosis de 1.125mg (3 comprimidos) cada 12 horas. En cualquier caso, siempre que se utilice efavirenz la dosis recomendada es de 1.125mg (3 comprimidos) cada 8 horas.
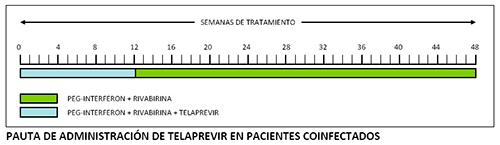
La duración total del tratamiento es de 48 semanas. Telaprevir debe administrarse en combinación con P-IFN alfa (2a o 2b) y RBV durante 12 semanas, seguidas de 36 semanas de tratamiento con P- IFN y RBV hasta completar las 48 semanas totales de tratamiento
A diferencia de lo que ocurre con boceprevir, la mayor parte del desarrollo clínico de telaprevir se ha realizado utilizando P-INF alfa 2a.
CRITERIOS DE SUSPENSIÓN DEL TRATAMIENTO EN PACIENTES COINFECTADOS POR VIH Y VHC
Para boceprevir
(1) Determinación del ARN-VHC en la semana 12 de tratamiento (semana 8 de triple terapia), y si es mayor o igual a 100 UI/ml todos los tratamientos (boceprevir, P-IFN y RBV) deben ser suspendidos.
(2) Determinación del ARN-VHC en la semana 24 y 36 de tratamiento (semana 20 y 32, respectivamente, de triple terapia), y si es detectable todos los tratamientos (boceprevir, P-IFN y RBV) deben ser suspendidos (esta decisión debe de basarse en una prueba con un límite inferior de detección de 10-15 UI).
(3) Si por cualquier razón se tuviera que suspender el P-IFN y la RBV, también se debe suspender el boceprevir.
Para telaprevir
(1) Determinación del RNA-VHC en la semana 4 de tratamiento, y si es superior a 1.000 UI/ml todos los tratamientos (telaprevir, P- IFN y RBV) deben ser suspendidos.
(2) Determinación del RNA-VHC en la semana 12 de tratamiento, y si es superior a 1000 UI/ml todos los tratamientos (P- IFN y RBV) deben ser suspendidos.
(3) Determinación del RNA-VHC en la semana 24 y 36 de tratamiento, y si es detectable todos los tratamientos (P-IFN y RBV) deben ser suspendidos.
(4) Si por cualquier razón se tuviera que suspender el P-IFN y la RBV, también se debe suspender el telaprevir
2. CRITERIOS DE USO EN PACIENTES CON TRASPLANTE HEPÁTICO Y VHC
Los pacientes trasplantados de hígado con recurrencia de la hepatitis por el VHC candidatos a triple terapia con P-INF, RBV y un inhibidor de la proteasa del VHC (boceprevir o telaprevir) han de cumplir todos los siguientes criterios de inclusión y ninguno de los de exclusión:
Criterios de inclusión
(1) Infección por VHC genotipo 1
(2) Fibrosis significativa o cirrosis (F2-F4) o hepatitis colestásica o colestásica fibrosante confirmada por biopsia hepática. A pesar de la fiabilidad de la elasticidad hepática para identificar el estadio de la fibrosis en pacientes trasplantados con recurrencia de la hepatitis C, la realización de una biopsia hepática es un requisito indispensable antes de iniciar el tratamiento.
(3) Concentración de hemoglobina >10,5 g/dl en mujeres y >11 g/dl en hombres
(4) Inmunosupresión con ciclosporina o tacrolimus (y prednisona a dosis inferiores o iguales a 10 mg/día). Los datos de interacción farmacológica de telaprevir/boceprevir con everolimus o rapamicina son limitados por lo que su uso debería restringirse a situaciones especiales (ver abajo).
Criterios de exclusión
(1) Cualquier contraindicación al tratamiento con interferón
(2) Presencia de rechazo o de hepatitis autoinmune en la biopsia hepática realizada antes de iniciar el tratamiento antiviral.
(3) Presencia de descompensación en el momento de indicar el tratamiento (ascitis, encefalopatía, hemorragia, ictericia).
(4) No existen suficientes datos sobre la incidencia de complicaciones graves en pacientes con cirrosis establecida tras un trasplante hepático en tratamiento con inhibidores de proteasa. En pacientes inmunocompetentes la presencia de plaquetopenia (<100.000 plaquetas/mm3) y de una albúmina <3,5 g/dl se asocian a una incidencia elevada de efectos adversos graves. Aunque en el contexto del trasplante estas variables pudieran no tener el mismo valor predictivo, sería recomendable aplicar estos mismos criterios en el contexto de la cirrosis post-trasplante.
(5) Infección activa (ya sea bacteriana, por citomegalovirus o fúngica)
(6) Insuficiencia renal (filtrado glomerular estimado inferior a 50 ml/min).
(7) Historia de complicaciones neurológicas relacionadas con el tratamiento inmunosupresor (convulsiones, encefalopatía, etc.)
DOSIS Y PAUTAS DE ADMINISTRACIÓN EN PACIENTES VHC TRASPLANTADOS
El tratamiento con ambos medicamentos debe ser iniciado y supervisado por un médico con experiencia en el manejo de pacientes trasplantados de hígado y de la hepatitis crónica C.
Boceprevir
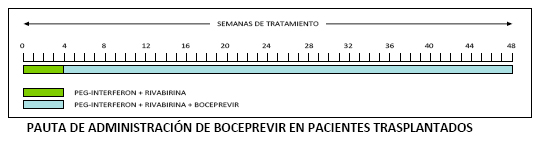
La dosis habitual de boceprevir es de 800mg (4 cápsulas) cada 8 horas (se recomienda intervalo de 7 a 9 horas), administrados con comida. Las cápsulas deben tragarse enteras. La duración total del tratamiento es de 48 semanas. Boceprevir debe administrarse en combinación con P-IFN alfa (2a o 2b) y RBV, iniciándose después de un periodo inicial de 4 semanas («lead-in») sólo con P-IFN y RBV. La triple terapia (boceprevir, P-IFN y RBV) debe mantenerse durante 44 semanas más hasta completar las 48 semanas totales de tratamiento.
Telaprevir
La dosis habitual de telaprevir es de 750mg (2 comprimidos) cada 8 horas, administrados con comida. Los comprimidos deben tragarse enteros (sin masticar o triturar o disolver). Aunque no existen datos en pacientes coinfectados y trasplantados, telaprevir se podrá administrar también a dosis de 1.125mg (3 comprimidos) cada 12 horas.
La duración total del tratamiento es de 48 semanas. Telaprevir debe administrarse en combinación con P-IFN alfa (2a o 2b) y RBV durante 12 semanas, seguidas de 36 semanas de tratamiento con P- IFN y RBV hasta completar las 48 semanas totales de tratamiento.

CRITERIOS DE SUSPENSIÓN DEL TRATAMIENTO EN PACIENTES CON HCC TRASPLANTADOS
Para boceprevir
(1) Determinación del ARN-VHC en la semana 12 de tratamiento (semana 8 de la triple terapia), y si es mayor o igual a 100 UI/ml todos los tratamientos (boceprevir, P-IFN y RBV) deben ser suspendidos.
(2) Determinación del ARN-VHC en la semana 24 y 36 de tratamiento (semana 20 y 32, respectivamente, de triple terapia), y si es detectable todos los tratamientos (boceprevir, P-IFN y RBV) deben ser suspendidos.
(3) Si por cualquier razón se tuviera que suspender el P-IFN y la RBV, también se debe suspender el boceprevir.
Para telaprevir
(1) Determinación del RNA-VHC en la semana 4 de tratamiento, y si es superior a 1.000 UI/ml todos los tratamientos (telaprevir, P- IFN y RBV) deben ser suspendidos.
(2) Determinación del RNA-VHC en la semana 12 de tratamiento, y si es superior a 1000 UI/ml todos los tratamientos (telaprevir, P-IFN y RBV) deben ser suspendidos.
(3) Determinación del RNA-VHC en la semana 24 y 36 de tratamiento, y si es detectable todos los tratamientos (P-IFN y RBV) deben ser suspendidos.
(4) Si por cualquier razón se tuviera que suspender el P-IFN y la RBV, también se debe suspender el telaprevir.
MANEJO DE LA CICLOSPORINA (CSA) Y TACROLIMUS (TAC) [1]
Dadas las interacciones farmacológicas entre los inhibidores de la proteasa y los inmunosupresores, se recomienda:
(1) En el caso de tratamiento con boceprevir: la dosis de CsA se debe reducir a la mitad el día del inicio del tratamiento antiviral (por ejemplo, un paciente con una dosis de CsA de 100mg/12 horas - dosis total diaria de 200mg- debe reducir la dosis a 100mg/24 horas). En cuanto a tacrolimus, se debe reducir la dosis 1/8 el día del inicio del tratamiento con boceprevir (por ejemplo, un paciente con una dosis de 2mg/12 horas –dosis total diaria de 4mg/día- debe reducir la dosis a 0,5mg/24 horas). Se puede también considerar también un ajuste de dosis espaciando la toma.
(2) En el caso de tratamiento con telaprevir, la dosis de CsA se debe reducir a una cuarta parte el día del inicio del tratamiento con telaprevir (por ejemplo, un paciente con una dosis de CsA de 100mg/12 horas –dosis total diaria de 200mg/día- debe reducir la dosis a 50mg/día). En cuanto a tacrolimus, la dosis se debe reducir a un 1/30 el día del inicio del tratamiento con telaprevir (por ejemplo, un paciente con una dosis de 1mg/12 horas -dosis total diaria de 2mg/día- debe reducir la dosis a 0,2mg/72 horas). La dosis de 0,2mg está comercializada (formulación pediátrica). Se puede también considerar ajuste de dosis espaciando la toma (en el ejemplo anterior el paciente debería tomar 0,5mg/semana).
(3) Dado que los cambios en las dosis e intervalos de CsA o TAC recomendados en los apartados anteriores son aproximaciones, se deben monitorizar los niveles valle de CsA o TAC en días alternos durante la primera semana de tratamiento con la triple terapia, realizando los ajustes de dosis necesarios para alcanzar los niveles deseados.
(4) En el caso de pacientes con recidiva grave e inmunosupresión no estable, el manejo de la CsA en el contexto de inhibidores de la proteasa es menos complejo que el del TAC y por tanto, sería recomendable cambiar a CsA.
(5) Una vez interrumpido el tratamiento con telaprevir o boceprevir se reiniciará el tratamiento inmunosupresor a las dosis previas al inicio de los inhibidores de la proteasa (y se procederá a monitorizar los niveles a días alternos durante la primera semana y semanalmente durante el primer mes). Una proporción significativa de pacientes requerirán dosis superiores a las basales tras la suspensión del inhibidor de la proteasa, especialmente si estaban recibiendo tacrolimus.
3. CARACTERÍSTICAS DE LOS CENTROS
La incorporación de telaprevir o boceprevir al tratamiento estándar de la HCC plantea problemas nuevos, como es el manejo de los efectos adversos inducidos por estos medicamentos, y el acceso rápido a los resultados de las pruebas de laboratorio necesarias para actuar con racionalidad y eficiencia. Prolongar indebidamente el tratamiento, cuando no se produce respuesta, incrementa la aparición de mutaciones de resistencia en el virus e incrementa innecesariamente el coste.
El tratamiento con triple terapia debe realizarse en unidades que cumplan una serie de requisitos mínimos para la óptima vigilancia de la seguridad de los pacientes. Es esencial que los pacientes sean tratados en el contexto de unidades que estén acostumbradas a la detección y manejo de las potenciales complicaciones. La idoneidad de los centros resulta particularmente relevante en los pacientes F4 con enfermedad muy avanzada. La posibilidad de consulta con especialistas que puedan abordar el manejo de problemas extra- hepáticos, como depresión o manifestaciones cutáneas resulta imprescindible.
Estos tratamientos exigen unas reglas de suspensión muy estrictas que están reguladas por la determinación del ARN-VHC, que se debe realizar de manera muy frecuente. Cualquier centro o unidad que utilice estos fármacos debe disponer de los resultados de esta determinación en menos de una semana (máximo 7 días).
Igualmente, tienen que poder disponer del resultado del polimorfismo IL28B, realizado en el laboratorio del propio hospital o en un laboratorio externo que pueda hacer esta determinación de manera regular y rápida.
Los centros que no cumplan estos requisitos tendrían que poder remitir a los pacientes que consideren potencialmente tributarios de recibir triple terapia a otro centro.
4. ENSAYOS CLÍNICOS AUTORIZADOS EN ESPAÑA CON BOCEPREVIR, TELAPREVIR U OTROS MEDICAMENTOS PARA EL TRATAMIENTO DE LA HEPATITIS C EN COINFECTADOS Y TRASPLANTADOS
Puede consultarse un listado completo y actualizado de los ensayos clínicos autorizados en España para el tratamiento de la hepatitis C crónica en pacientes coinfectados por VIH y en pacientes con trasplante hepático en la dirección http://www.aemps.gob.es. Desde enero de 2013 esta misma información está disponible también en el Registro Español de estudios clínicos, en la dirección https://reec.aemps.es/
5. NOTIFICACIÓN DE REACCIONES ADVERSAS
Se recuerda la importancia de notificar todas las sospechas de reacciones adversas a los Centros Autonómicos de Farmacovigilancia, pudiéndose realizar a través de la web https://www.notificaram.es
Estos medicamentos son de reciente comercialización y están sujetos a un seguimiento adicional por lo que la notificación de sospechas de reacciones adversas es prioritaria.
6. RECOMENDACIONES PARA EL MANEJO DEL EXANTEMA
El manejo y tratamiento de la toxicidad cutánea debe llevarse a cabo en base a la práctica clínica habitual, recomendándose adicionalmente los siguientes procedimientos para la evaluación y manejo de la misma:
Evaluación del grado de severidad
Todos los acontecimientos cutáneos se evaluarán junto con otros síntomas sistémicos y anomalías analíticas.
El grado y la severidad de la lesión deben ser evaluados utilizando los criterios descritos a continuación:
(1) Grado 1, leve: el exantema leve se define como una erupción cutánea localizada o una erupción cutánea con una distribución limitada (por ejemplo, en varias localizaciones aisladas del cuerpo), con o sin prurito acompañante. Un exantema leve no tendrá lesiones en escarapela, signos sistémicos ni afectación de las mucosas ni signos de desprendimiento epidérmico.
(2) Grado 2, moderado: el exantema moderado se define como una erupción cutánea difusa que afecta, como máximo, al 50% de la superficie corporal, con o sin separación de la piel superficial o prurito. Puede haber inflamación de las mucosas sin úlceras. Obsérvese que los trastornos de las mucosas que no estén relacionados con episodios cutáneos, como úlceras aftosas, estomatitis o liquen plano bucal, deben quedar bien documentados y no se tienen en cuenta para graduar el exantema. Un exantema moderado no tendrá signos de lesiones en escarapela ni desprendimiento epidérmico. Puede haber signos y síntomas sistémicos moderados, como fiebre, artralgia o eosinofilia, con un exantema morbiliforme moderado. Por consiguiente, para la gradación del exantema se necesitan el tipo de éste y una evaluación minuciosa del grado de todo signo sistémico y su relación temporal con el inicio del exantema o su progresión.
(3) Grado 3, intenso: el exantema intenso se define como un exantema generalizado que afecta a más del 50% de la superficie corporal o que presenta alguna de las características siguientes:
- exantema con vesículas o ampollas;
- úlceras superficiales en las mucosas;
- desprendimiento epidérmico (necrosis de todo el espesor epidérmico y separación de la epidermis de la dermis subyacente);
- lesiones en escarapela atípicas o típicas; <
- púrpura palpable o eritema que no blanquea a la presión;
- diagnóstico de eosinofilia farmacológica con síntomas sistémicos (DRESS), eritema multiforme (EM) o pustulosis exantemática generalizada aguda (PEGA).
- Un exantema con presencia de signos o síntomas sistémicos importantes que sean nuevos y se consideren relacionados con el inicio o la progresión del mismo se considerará de grado 3.
(4) Grado 4, potencialmente mortal: Diagnóstico de erupción ampollosa generalizada, síndrome de Stevens-Johnson (SSJ) o necrolisis epidérmica tóxica (NET).
Recomendaciones profilácticas
Se debe recomendar a los pacientes el uso diario de cremas hidratantes y emolientes.
Recomendaciones de tratamiento
Los antihistamínicos y corticosteroides tópicos pueden proporcionar alivio sintomático a los pacientes que presenten exantema y síntomas acompañantes, como prurito.
El tratamiento del exantema durante el periodo de tratamiento debe ajustarse siempre a la práctica clínica habitual.
Entre los antihistamínicos tópicos y sistémicos recomendados en todos los grados de exantema figuran difenhidramina, hidroxizina, levocetirizina y desloratadina. Deberá realizarse un seguimiento sintomático de los pacientes para detectar un posible aumento de la exposición y empeoramiento de acontecimientos adversos.
Dado que la inmunodepresión debida a los corticosteroides sistémicos puede causar elevaciones del ARN del VHC y debido a la sospecha de interacciones farmacológicas, éstos sólo deberán utilizarse cuando sea clínicamente necesario y tras haber empleado otros tratamientos y medidas. Se permitirá el uso de corticosteroides tópicos, aunque deberá limitarse a períodos breves, p. ej., un máximo de 2 semanas de uso continuo o regular y limitado al 50% de la superficie corporal como máximo. En esta situación se recomendará encarecidamente el uso de cremas o lociones, por su menor potencial de absorción. Se desaconseja el uso de geles o pomadas de corticosteroides tópicos, pues su potencial de absorción es relativamente mayor.
Los pacientes que presenten un exantema (de cualquier grado) que requiera tratamiento con corticosteroides sistémicos deberán interrumpir el tratamiento de manera inmediata y definitiva.
Recomendaciones de tratamiento en los pacientes que presenten un exantema de grado 1 ó 2.
El seguimiento se llevará a cabo bajo criterio del médico responsable de la prescripción. Podrán emplearse medicamentos para aliviar los síntomas según lo descrito anteriormente. Además, a los pacientes que manifiesten exantema o prurito cutáneo se les recomendarán otras estrategias para reducir al mínimo la intensidad o progresión de sus signos y síntomas (p. ej., limitar la exposición al sol y el calor, baños con bicarbonato sódico o avena o prendas de vestir poco ajustadas). En todos los casos se vigilará de cerca a los pacientes para detectar la progresión del exantema o el empeoramiento de los signos y síntomas de afectación sistémica y se realizará un seguimiento hasta la resolución completa del exantema.
En general, no será necesario interrumpir el tratamiento en los pacientes que presenten un exantema grado 1, mientras que en los que presenten un exantema de grado 2 que progrese o no mejore, se considerará la conveniencia de esa interrupción. Si es preciso interrumpir el tratamiento por un exantema, se interrumpirá en primer lugar el inhibidor de la proteasa, si el exantema no mejora sintomática ni objetivamente en el plazo de 7 días después de interrupción, se interrumpirá el uso de RBV.
La administración de P-IFN alfa podrá mantenerse, a menos que también esté médicamente indicada su interrupción.
La administración de los inhibidores de la proteasa no podrá reanudarse después de haberla suspendido a consecuencia de un acontecimiento adverso. Si se suspende P-IFN alfa o RBV por un exantema, podrá reanudarse si éste mejora en los 14 días siguientes a la interrupción del medicamento respectivo.
Recomendaciones de tratamiento en los pacientes que presenten un exantema de grado 3
En los pacientes que presenten un exantema de grado 3 se aplicarán las mismas recomendaciones generales dadas para el exantema de grado 1 y 2.
En los pacientes que presenten un exantema de grado 3, deberá interrumpirse el uso de los inhibidores de la proteasa inmediata y definitivamente. Si el exantema no mejora sintomática ni objetivamente en el plazo de 7 días después de su interrupción, se deberá considerar la interrupción o discontinuación del tratamiento con RBV y/o P-INF alfa de forma secuencial o simultánea. Si se considera médicamente indicado, podría ser necesaria una interrupción o discontinuación de RBV y peg-interferón alfa de forma más temprana. Se podrán interrumpir los tres fármacos simultáneamente si está clínicamente indicado. En todo caso, los pacientes que presenten un exantema grave progresivo o aquellos con diagnóstico o sospecha de DRESS, EM o PEGA interrumpirán todo el tratamiento de manera inmediata y definitiva.
La administración de los inhibidores de la proteasa no podrá reanudarse después de haberla suspendido a consecuencia de un acontecimiento adverso. Si se suspende peg-IFN alfa o RBV por un exantema, podrá reanudarse si éste mejora en los 14 días siguientes a la interrupción del medicamento respectivo. Si no es posible su reintroducción, debería suspenderse todo el tratamiento pues la monoterapia no está justificada en estos pacientes. Los pacientes que suspendan el tratamiento por DRESS, EM o PEGA no deberán reanudar la toma de ninguno de los tres fármacos.
Se recomienda la derivación a un dermatólogo en caso de exantema de grado 3.
Se recomienda la realización de controles analíticos que incluyan: hemograma (con fórmula), ALT, AST y creatinina. Se realizarán otros estudios analíticos según proceda.
Se instaurará un seguimiento clínico estrecho y la intervención médica oportuna. Es posible que se requiera un seguimiento diario, para vigilar la progresión del acontecimiento desde su aparición hasta que se observe mejoría. Se vigilará a todos los pacientes hasta la resolución completa del exantema.
Recomendaciones de tratamiento en los pacientes que presenten un exantema de grado 4
En los pacientes que presenten un exantema de grado 4 se aplicarán las mismas recomendaciones generales dadas para un exantema de grado 1, 2 y 3.
Los pacientes con diagnóstico o sospecha de un exantema que se considere potencialmente mortal (exantema de grado 4), incluidos el SSJ y NET, interrumpirán todo el tratamiento de manera inmediata y definitiva. Tal y como se indica en la Ficha Técnica de telaprevir, durante la experiencia postcomercialización se han notificado casos de NET con desenlace mortal.
Se recomienda la derivación a un dermatólogo como en el caso anterior.
Se recomienda la realización de controles analíticos que incluyan: hemograma (con fórmula), ALT, AST y creatinina. Se realizarán otros estudios analíticos según proceda.
Se instaurará un seguimiento clínico estrecho y la intervención médica oportuna. Es posible que se requiera un seguimiento diario, para vigilar la progresión del acontecimiento desde su aparición hasta que se observe mejoría. Se vigilará a todos los pacientes hasta la resolución completa del exantema.
7. RECOMENDACIONES PARA EL MANEJO DE LA ANEMIA SECUNDARIA AL TRATAMIENTO
En primer lugar, la estrategia recomendada para el manejo de la anemia inducida por el tratamiento es la reducción de las dosis de ribavirina. Los cambios en la dosis de ribavirina deben hacerse basándose en las recomendaciones de la ficha técnica del producto. Debido a la limitada experiencia clínica con el uso de ribavirina a dosis inferiores a 600mg al día, no se recomiendan reducciones de dosis por debajo de ésta en la medida de lo posible. La reducción de la dosis de ribavirina debe hacerse con especial precaución en pacientes con cirrosis ya que la evidencia del impacto en la tasa de curación es más escasa. Debe tenerse en cuenta que si para el tratamiento de la anemia se decide la suspensión permanente de ribavirina, los inhibidores de la proteasa deben suspenderse de forma permanente.
Otras intervenciones para el manejo de la anemia, incluyendo el uso de agentes estimulantes de la eritropoyesis (AES) y la trasfusión de concentrado de hematíes, pueden ser necesarias si las medidas anteriores son insuficientes. No obstante, deben tenerse en cuenta el riesgo de eventos trombóticos asociados al uso de AES en estos pacientes, así como las advertencias específicas de la AEMPS sobre el riesgo de aplasia eritrocítica pura, mediada por anticuerpos anti- eritropoyetina, y proliferación tumoral.
REFERENCIAS
1. Coilly A, Roche B, Samuel D. Current management and perspectives for HCV recurrence after liver transplantation. Liver International (2013) 33 (1): 56–62
GRUPO DE EXPERTOS
(por orden alfabético)
Juan Berenguer Berenguer
Unidad de Enfermedades Infecciosas/VIH. Hospital General Universitario Gregorio Marañón de Madrid
José Luis Calleja Panero
Servicio de Gastroenterología (Unidad de Hepatología). Hospital Universitario Puerta de Hierro Majadahonda
Rafael Esteban Mur
Servicio de Hepatología. Hospital Vall d'Hebron, Barcelona
Mª Jesús Fernández Cortizo
División de Farmacología y Evaluación Clínica. Agencia Española de Medicamentos y Productos Sanitarios
Xavier Forns Bernhardt
Servicio de Hepatología. Hospital Clinic de Barcelona, Ciberehd, IDIBAPS
Juan González García
Servicio de Medicina Interna (Unidad de VIH). Hospital Universitario La Paz de Madrid
Antonio López Navas
División de Farmacología y Evaluación Clínica. Agencia Española de Medicamentos y Productos Sanitarios
Luis Margusino Framiñán
Servicio de Farmacia. Hospital A Coruña, Complexo Hospitalario Universitario A Coruña.
Ramón Morillo Verdugo
Servicio de Farmacia. Hospital de Valme, Sevilla
Óscar Pinar López
Subdirección de Compras de Farmacia y Productos Sanitarios. Servicio Madrileño de Salud
Manuel Romero Gómez
Unidad de Gestión Médico-Quirúrgica de Enfermedades Digestivas y Ciberehd. Hospital Universitario de Valme. Universidad de Sevilla
Belén Ruiz Antorán
Servicio de Farmacología Clínica. Hospital Universitario Puerta de Hierro Majadahonda
Arantxa Sancho López
Comité de Medicamentos de Uso Humano (CHMP) de la EMA; Agencia Española de Medicamentos y Productos Sanitarios
Maria Jesús Téllez Molina
Servicio de Medicina Interna (Unidad de VIH). Hospital Clínico San Carlos de Madrid
Miguel Angel von Wichmann de Miguel
Unidad de Enfermedades Infecciosas. Hospital Donostia (San Sebastián)
Todos los expertos han realizado una declaración de conflictos de interés.
Este documento ha sido aprobado por el Grupo de Coordinación del posicionamiento Terapéutico, que ha elevado, a su vez, un documento de revisión de la estrategia terapéutica recomendada para el uso de inhibidores de la proteasa en el tratamiento de la hepatitis C crónica (VHC) a la Comisión Permanente de Farmacia.
Fuente: Agencia Española de Medicamentos y Productos Sanitarios.
