Informe de Posicionamiento Terapéutico de Viekirax® (ombitasvir/paritaprevir/ritonavir) y Exviera® (dasabuvir)
PT-OMB/PARITA/RITO_DASA/V1/20032015
Informe de Posicionamiento Terapéutico de Viekirax®(ombitasvir/paritaprevir/ritonavir) y Exviera® (dasabuvir)
Fecha de publicación: 20 de marzo de 2015
Fecha de corrección: 14 de abril de 2015 (ver al final)
La infección por el virus C de la hepatitis (VHC) es un problema de salud de primera magnitud en España, con una prevalencia aproximada del 1,5% (virémicos 1%; rango 0,8-1,3). La infección crónica es responsable de unas 4.000 muertes al año y supone la primera causa de trasplante hepático.
El virus C está dividido en 7 genotipos con numerosos subtipos, siendo el más frecuente en nuestro país el 1b (44%), seguido por 1a (26%), 3 (20%), 4 (8%) y 2 (3%). Aunque el genotipo no influye claramente en la progresión de la enfermedad sí que supone un factor de respuesta con los regímenes terapéuticos actuales. El tratamiento del virus C durante los últimos años ha estado basado en el uso de interferón (IFN) y ribavirina (RBV), siendo posible desde 2011 asociar, en los pacientes con genotipo 1, los primeros inhibidores de la proteasa NS3/4A (boceprevir y telaprevir). Los tratamientos basados en interferón se asocian a un gran número de efectos secundarios, alguno de ellos graves. Los pacientes con cirrosis están especialmente expuestos a estos efectos graves e, incluso, a mortalidad.
En los últimos años hemos asistido a un rápido desarrollo de nuevos medicamentos frente al virus C, tales como sofosbuvir, simeprevir, daclatasvir y ledipasvir encontrándose varias moléculas más en distintas fases de estudio.
OMBITASVIR/PARITAPREVIR /RITONAVIR (VIEKIRAX®) Y DASABUVIR (EXVIERA®)
Viekirax®(OBV/PTV/rtv) y Exviera®(DSV) han sido autorizados, combinados entre sí o con otros medicamentos, en el tratamiento de la hepatitis C. En el momento actual, la pauta habitual de combinación es OBV/PTV/rtv ± DSV ± RBV. Los medicamentos Viekirax® y Exviera® no deben utilizarse en monoterapia. En su desarrollo se realizaron pequeños estudios en combinación con interferón pegilado. Se está realizando, al menos, un estudio en combinación con sofosbuvir (SOF) en pacientes con fallo previo a otros AADs. No existen estudios de combinación con telaprevir, boceprevir, simeprevir, daclatasvir ni otras moléculas similares.
Farmacología
Viekirax® combina dos antivirales con mecanismos de acción diferenciados y perfiles de resistencia no solapados, actuando contra diferentes puntos del ciclo replicativo de los genotipos 1 y 4 del virus C. Este efecto se ve potenciado por la asociación, en algunos pacientes, con DSV.
Cada comprimido de Viekirax® está compuesto por ombitasvir (12,5 mg), paritaprevir (75 mg) y ritonavir (50 mg). La dosis recomendada es de 2 comprimidos, una vez al día, tomados con mg de dasabuvir. La dosis recomendada es de 1 comprimido cada 12 horas, tomado con alimento. Ver Tabla 1.
Tabla 1: Duración recomendada de tratamiento con Viekirax®/Exviera® y el uso recomendado de añadir ribavirina en determinados subgrupos.
| Población Pacientes* | Tratamiento* | Duración |
| Genotipo 1b, sin cirrosis | OBV/PTV/rtv + DSV | 12 semanas |
| Genotipo 1b, con cirrosis compensada | OBV/PTV/rtv + DSV + RBV | 12 semanas |
| Genotipo 1a, sin cirrosis | OBV/PTV/rtv + DSV + RBV | 12 semanas |
| Genotipo 1a, con cirrosis compensada | OBV/PTV/rtv + DSV + RBV | 24 semanas |
| Genotipo 4, sin cirrosis | OBV/PTV/rtv + RBV | 12 semanas |
| Genotipo 4, con cirrosis compensada | OBV/PTV/rtv + RBV | 24 semanas |
* Incluye pacientes co-infectados con el Virus de la Inmunodeficiencia Humana (VIH).
Ritonavir (rtv) : es un inhibidor del citocromo CYP3A que aumenta la exposición de su sustrato paritaprevir. Ritonavir carece de actividad frente al virus C.
Ombitasvir (OBV, ABT-267) : es un inhibidor de la proteína NS5A, la cual es fundamental para la replicación del virus C. Según los resultados del modelo replicón, OBV pudiera haber sido útil en el tratamiento del genotipo 3 pero sólo estará disponible en asociación con PTV, el cual tiene una actividad significativamente reducida frente a este genotipo. Su mecanismo de eliminación es biliar, recuperándose un 90% de la dosis en las heces y tan sólo un 2% en la orina. Su principal metabolismo es a través de CYP3A4. Su concentración aumenta 2,2 veces en la insuficiencia hepática severa.
Paritaprevir (PTV, ABT-450) : es un inhibidor de la proteasa no estructural (NS)3/4A, la cual es fundamental para la replicación del virus C. Administrado con rtv (PTV/rtv) se elimina en un 88% por las heces y en un 8% por la orina. Principalmente, se metaboliza a través de CYP3A4/5. Su concentración aumenta un 62% en la insuficiencia hepática moderada y 9 veces en la severa.
Dasabuvir (DSV, ABT-333) : es un inhibidor no nucleósido de la polimerasa NS5B. Su mecanismo de eliminación es biliar (95%), eliminándose tan sólo en un 2% por la orina. Su catabolismo es a través de CYP2C8 y CYP3A4. La insuficiencia hepática leve, moderada o grave afecta a su concentración en un aumento del 17%, disminución del 16% y aumento del 325%, respectivamente. Los cambios en la concentración de DSV en insuficiencia hepática leve y moderada no se consideran clínicamente relevantes.
Eficacia
El desarrollo clínico de estos fármacos consta de múltiples estudios que incluyen un gran número de pacientes infectados por el VHC genotipo 1. A continuación se resumen los más relevantes.
1. ESTUDIO M11-652 (AVIATOR)
El principal ensayo que determinó la selección de los regímenes para la fase III fue el estudio M11-652 (estudio multicéntrico, abierto, de fase II, que evalúa distintos regímenes de OBV, PTV/rtv y DSV ± RBV en pacientes con hepatitis C crónica (HCC) infectados con el Genotipo 1 del VHC sin cirrosis). Incluyó 567 pacientes (55% hombres, edad media 50,4 años, 66% G1a, 34% con estadio de fibrosis ≥2) divididos en 2 cohortes (cohorte 1: 438 pacientes naïve; cohorte 2: 133 pacientes respondedores nulos a interferón pegilado (PEG) y RBV) en los que se combinaban diversas dosis de PTV/rtv, asociado a OBV, RBV y/o DSV (14 brazos en total), y con una duración de los tratamientos de 8, 12 ó 24 semanas (Figura 1).
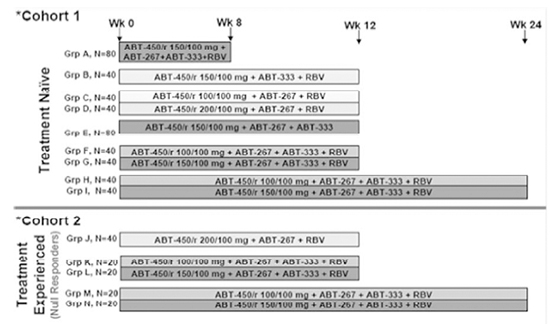
Fig. 1: Diseño del estudio M11-652 (AVIATOR).
El seguimiento post-tratamiento fue de 48 semanas. El elevado número de pacientes, inhabitual en ensayos fase II, permitió estudiar múltiples opciones terapéuticas. No existía ningún brazo de PTV/r + OBV sin RBV o sin DSV. Los resultados globales en las respuestas virológicas sostenidas tras 24 semanas de seguimiento post- tratamiento (RVS24) fueron de un 89,7% en la cohorte 1 (naïve) y 92,5% en la cohorte 2 (respondedores nulos). Los resultados por subgrupos se exponen en la Tabla 2. En todos los grupos, los criterios tradicionales de mala respuesta (IL28B no-CC, elevada viremia, raza negra) carecieron de utilidad.
Tabla 2: RVS12 por subgrupos y subgenotipos 1 (ITT) en estudio AVIATOR
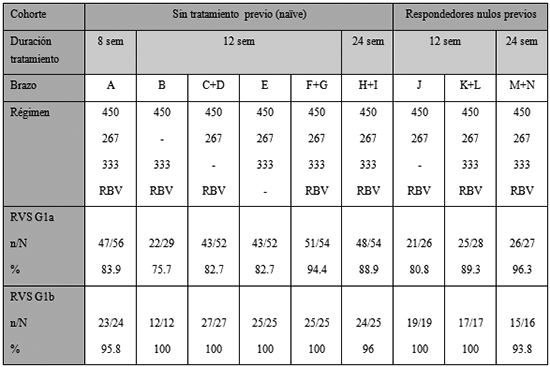
Este estudio permitió obtener la información necesaria sobre las combinaciones de OBV/PTV/rtv y DSV, con o sin RBV, y la duración de las mismas que, posteriormente, serían evaluadas en los ensayos fase III. En pacientes no cirróticos G1, 8 semanas de tratamiento resultan subóptimas y más de 12 no aumentan la eficacia. No obstante, se apreciaba que el tratamiento óptimo puede diferir entre genotipos. En G1a, eliminar del tratamiento OBV, DSV o RBV da como resultado menores RVS (<85%), mientras que la combinación OBV/PTV/rtv y DSV + RBV resultaba en RVS del 94,4% en pacientes naïve. En pacientes G1b, el beneficio de la adición a PTV/rtv de un tercer medicamento, sea OBV, DSV o RBV, es menos claro teniendo en cuenta los resultados en todos los brazos de G1b de este estudio (±99%). La combinación OBV/PTV/rtv y DSV con RBV durante 8 semanas en G1b obtuvo un 95,8% de RVS, de fase III.
A la vista de estos resultados, la compañía procedió a estudiar en sus ensayos fase III para G1 exclusivamente la combinación de 3AAD, con o sin RBV. No obstante, se debe destacar que entre los distintos regímenes y duraciones probadas y, especialmente en pacientes con VHC G1b, la combinación de OBV/PTV/rtv, + RBV, durante 12 semanas consigue tasas de RVS del 100% (27/27 pacientes) en pacientes naïve. En respondedores nulos, la tasa de RVS con OBV/PTV/rtv + RBV durante 12 semanas fue del 100% (19/19 pacientes).
2. ESTUDIOS PRINCIPALES
Existen 6 estudios principales de fase III que incluyen alrededor de 2300 pacientes con genotipo 1; 3 estudios en pacientes naïve sin cirrosis (SAPPHIRE-I, PEARL-III y PEARL-IV) y 2 estudios en pacientes pretratados con PEG/RBV sin cirrosis (SAPPHIRE-II y PEARL-II) y 1 en pacientes con cirrosis compensada que incluye tanto pacientes naïve como pacientes con tratamiento previo con PEG/RBV (TURQUOISE-II). Existen cuatro estudios adicionales: uno en trasplantados con fibrosis no avanzada (genotipo 1) y tres en pacientes sin tratamiento previo y pretratados (genotipo 4, genotipo 1 en tratamiento con agonistas opiáceos y coinfectados con VIH). Se resumen en la Tabla 3.
Tabla 3: Principales estudios con Viekirax y Exviera.
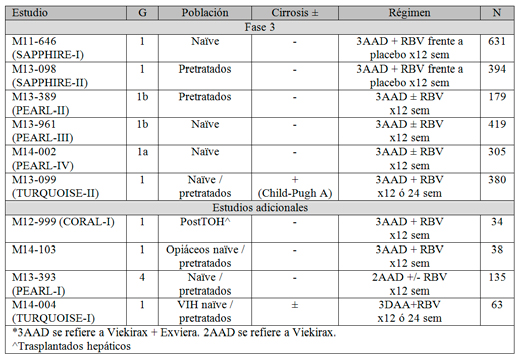
En los estudios SAPPHIRE I y II existía un brazo placebo cuya finalidad, asumiendo una tasa de 0 en RVS, era valorar los efectos adversos. Sin embargo, en todos los estudios, la eficacia se comparó con tasas de controles históricos (telaprevir más interferón más ribavirina) (tasas entre 47-78%), realizando una primer análisis de no inferioridad y, si esta se demostraba, se hacía un análisis de superioridad.. El margen establecido de no inferioridad fue 10,5%. Los estudios PEARL II, III y IV se diseñaron para evaluar la eficacia de la pauta 3AAD con o sin RBV. El objetivo principal es establecer la no inferioridad frente a un control histórico y, entre los objetivos secundarios, figura comparar la eficacia de añadir RBV a la pauta de tratamiento 3AAD frente a no añadirla. En los estudios que incluían pacientes no respondedores a tratamiento previo con PEG y RBV se utilizaron los criterios habituales de estratificación de no respuesta (recaedores, parciales, nulos); no se incluyeron pacientes sin respuesta a telaprevir o boceprevir ni a cualquier otro AAD. En todos los estudios, entre el 60-70% de los pacientes tenían grado de fibrosis F0-F1, un 15-20% F2 y 8-18% F3 según el estudio. No se incluyeron pacientes con grado de fibrosis F4, excepto en el estudio TURQUOISE-II, donde todos los pacientes eran cirróticos compensados. La existencia de cirrosis se estableció según biopsia hepática (METAVIR >3 o Ishak >4) o elastometría (Fibroscan) ≥14,6 kPa. La ausencia de cirrosis se estableció mediante biopsia hepática con resultado METAVIR ≤3 o Ishak ≤4, Fibroscan <9,6 kPa o Fibrotest ≤0,72 y APRI <2. La cifra mínima aceptable de plaquetas era, en cirrosis compensada, de 60.000/mL y, en no cirróticos, 120.000/mL. No se admitieron pacientes con descompensación previa. La RBV se dosificó a razón de 1.000 mg/día (<75 kg) o 1.200 mg/día (>75 kg). Las principales características demográficas de los estudios en fase III se recogen en la Tabla 4; las cifras corresponden a pacientes aleatorizados a tratamiento activo.
Tabla 4: Principales datos demográficos de los estudios fase III.
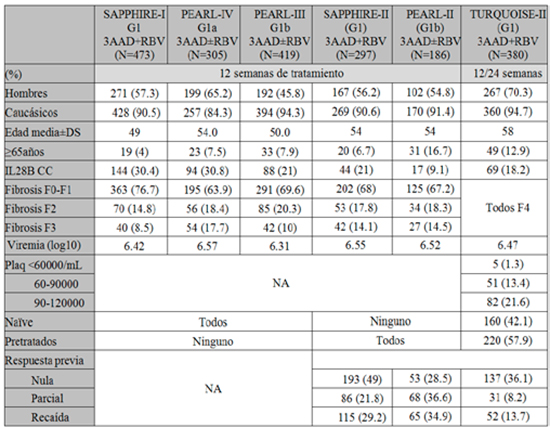
En todos los estudios la variable principal fue la respuesta viral sostenida en la semana 12 (RVS12), definida como ARN VHC indetectable o incuantificable (<25 UI mL) 12 semanas después de finalizar el tratamiento.
2.1. ADULTOS NAÏVE SIN CIRROSIS GENOTIPO 1
2.1.1. ESTUDIO SAPPHIRE-I (M11-646)
Estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo, donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con RBV durante 12 semanas en adultos con HCC genotipo 1 (68% G1a; 32% G1b).
Los pacientes se aleatorizaron para tratamiento con 3AAD + RBV o placebo (3:1) durante 12 semanas; los pacientes en el brazo placebo fueron posteriormente retratados de forma abierta. La tasa de RVS12 global fue del 96,2%. Por subgenotipos, los resultados fueron 95,3% en G1a (307/322) y 98% en G1b (148/151). 1 paciente presentó fallo virológico durante el tratamiento; 7 pacientes presentaron recaída post-tratamiento (6 con G1a); el resto de no respondedores se deben a pérdida de datos en semana 12 post- tratamiento o suspensión precoz de éste.
2.1.2. ESTUDIO PEARL-IV (M14-002)
Estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con y sin RBV durante 12 semanas en adultos con HCC genotipo 1a.
Los pacientes se aleatorizaron a tratamiento durante 12 semanas con 3AAD, con o sin RBV (1:2). El porcentaje global de RVS12 fue del 93% (97% con RBV, 90,2% sin RBV). Los principales motivos de no respuesta en el grupo sin RBV fueron recaída post-tratamiento (5,2%) y fallo virológico durante el tratamiento (2,9%).
2.1.3. ESTUDIO PEARL-III (M13-961)
Estudio multicéntrico, doble ciego, aleatorizado, donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con y sin RBV durante 12 semanas en adultos con HCC genotipo 1b.
Los pacientes se aleatorizaron a tratamiento durante 12 semanas con 3AAD, con o sin RBV (1:1). La RVS12 global fue del 99,3% (99,5% con RBV, 99% sin RBV). Dos de los tres pacientes no respondedores lo fueron por pérdida de datos. Hubo un fallo virológico durante el tratamiento en el brazo de tratamiento con RBV.
2.2. ADULTOS PREVIAMENTE TRATADOS SIN CIRROSIS, G1
2.2.1. ESTUDIO SAPPHIRE-II (M13-098)
Estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con RBV durante 12 semanas en adultos con previamente tratados con PEG/RBV con HCC genotipo 1 (59% G1a; 41% G1b).
Los pacientes se aleatorizaron a tratamiento durante 12 semanas con 3AAD + RBV o placebo (3:1). Los pacientes en el brazo placebo fueron posteriormente tratados de forma abierta. Los principales resultados se recogen en la Tabla 5.
Tabla 5: RVS12 y causas de no respuesta previa en el estudio SAPPHIRE-II.
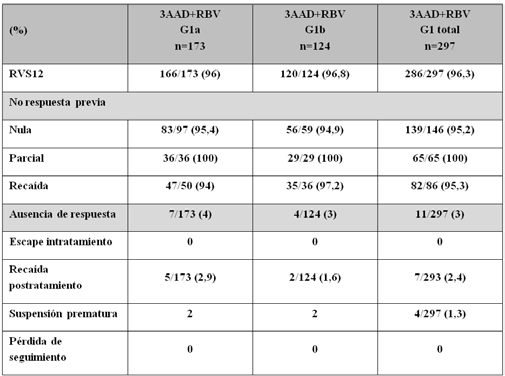
De los 7 pacientes con recaída post-tratamiento, 6 eran IL28B CT/TT y 3 presentaban un estadio 3 de fibrosis.
2.2.2. ESTUDIO PEARL II (M13-389)
Estudio multicéntrico, abierto, aleatorizado, donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con y sin RBV durante 12 semanas en adultos previamente tratados con HCC genotipo 1b.
Los pacientes se aleatorizaron a tratamiento durante 12 semanas con 3AAD con o sin RBV (1:1). La RVS12 global fue 98,3% (97,7% con RBV, 100% sin RBV). Las causas de no respuesta fueron pérdida de seguimiento y suspensión prematura del tratamiento.
Dado que en el ensayo AVIATOR la eficacia del tratamiento con 3AAD sin RBV resultó inferior en G1a, en la fase III, esta combinación en los pacientes sin respuesta previa a tratamiento sólo se estudió en G1b.
2.3. ADULTOS CON CIRROSIS COMPENSADA, G1
2.3.1. ESTUDIO TURQUOISE II (M13-099)
Estudio multicéntrico, abierto, aleatorizado, donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con RBV durante 12 ó 24 semanas en adultos con HCC genotipo 1 y cirrosis compensada (Child-Pugh A). No se admitieron pacientes con descompensación previa.
El objetivo principal era establecer la no inferioridad frente a un control histórico y entre los objetivos secundarios figura comparar la eficacia de las distintas duraciones de tratamiento.
Constituye el primer estudio de registro en el campo de la hepatitis C dedicado exclusivamente a pacientes con cirrosis compensada. Se incluyeron pacientes G1a y G1b, naïve o no respondedores a tratamiento previo con PEG/RBV. Los pacientes se aleatorizaron a tratamiento con 3AAD + RBV durante 12 ó 24 semanas.
La RVS en los pacientes tratados durante 12 y 24 semanas fue de 91,8% (191/208) y 95,9% (165/172), respectivamente. En pacientes G1b, la RVS fue alta independientemente de la duración del tratamiento (sólo 1/119 recaída tras 12 semanas de tratamiento). En pacientes G1a, la RVS fue >90% en todos los subgrupos tratados durante 12 y 24 semanas, exceptuando el caso de pacientes respondedores nulos tratados 12 semanas, en los que la tasa de RVS12 fue del 80% (ver Tabla 6)
Tabla 6: RVS12 en subgrupos de pacientes del estudio TURQUOISE II (M13-099). (ITT).
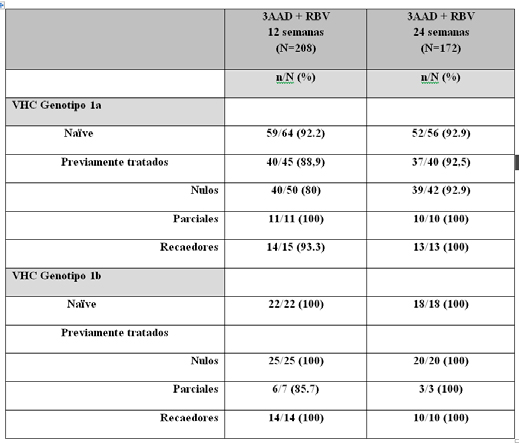
El fallo virológico durante el tratamiento sólo se produjo en 4 pacientes, todos ellos G1a (3/4 del grupo de 24 semanas de tratamiento).
En cuanto a las recaídas post-tratamiento, las tasas de recaída fueron superiores en las pautas de 12 semanas. En G1a hubo 11/140 (8%) en el brazo de 12 semanas de tratamiento y 1/121 (1%) en el de 24. En un intento de identificar a los pacientes G1a candidatos a tratamiento con 12 ó 24 semanas, se realizó un análisis de regresión logística de los factores de riesgo de recaída tras 12 semanas de tratamiento, identificándose como factores: sexo masculino, IL28B- TT y datos sugerentes de una cirrosis más avanzada (alfafetoproteína >20 ng/mL, albúmina <3,5 g/dL, <90.000 plaquetas/mL). Refiriéndonos específicamente al recuento de plaquetas, los pacientes con cifras menores (60.000-90.000/mL) presentaron peores resultados con 12 semanas de tratamiento (25/30, 83,3%) que con 24 (25/26, 96,2%); este efecto de la trombopenia no se observó cuando el recuento de plaquetas fue >90.000 plaquetas/mL (93,3% frente a 95,9%, respectivamente).
2.4. ADULTOS SIN CIRROSIS, G4
2.4.1. ESTUDIO PEARL-I (M13-393)
Estudio fase IIb, multicéntrico, abierto, aleatorizado, donde se evalúa la eficacia y seguridad de la combinación PTV/rtv y OBV con y sin RBV, durante 12 y 24 semanas, en un total de 8 brazos (pacientes con genotipo 4 y 1b, naive y pretratados con biterapia). Los grupos que incluyeron pacientes con genotipo 4 fueron los grupos: 1 (OBV/PTV/rtv 12 semanas en pacientes naïve), 4 (OBV/PTV/rtv con RBV 12 semanas en pacientes naïve) y 6 (OBV/PTV/rtv con RBV 12 semanas en pacientes pretratados). Todos ellos de pacientes no cirróticos. El grupo 5 no se abrió al estudio. (Figura 2).
Por otro lado, únicamente se incluyeron pacientes con cirrosis compensada genotipo 1b, correspondientes a los grupos 7 y 8, tratados con PTV/rtv y OBV, sin RBV, naïve o previamente tratados.
Los sujetos tratados (N=135) tenían una mediana de edad de 51 años (intervalo 19-70); 63,7 % eran naïve, 17% eran no respondedores al tratamiento previo PEG/RBV, 6,7% eran respondedores parciales al tratamiento previo PEG/RBV, 12,6% eran sujetos con recidiva previa durante un régimen de PEG/RBV, 65,2 % eran varones; 8,9 % eran de raza negra; 14,1 % tenían un índice de masa corporal de al menos 30 kg/m2; 69,6 % presentaban niveles basales de ARN del VHC de 800.000 UI/ml como mínimo; 78,5 % tenían el genotipo IL28B no-CC; 6,7 % tenían fibrosis avanzada (F3).
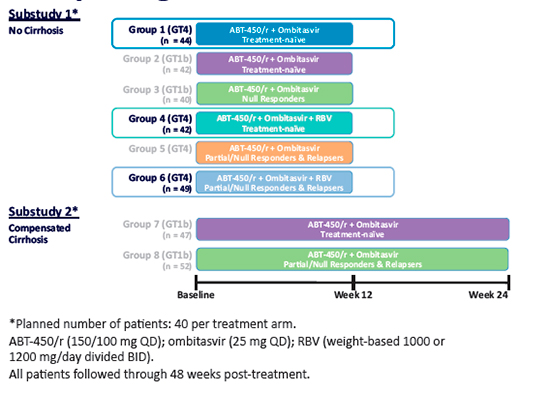
Fig. 2: Diseño del estudio PEARL-1 (M13-393).
Tanto los pacientes G4 naïve (42/42) como los no respondedores previamente a PegIFN/RBV (49/49) tratados con PTV/rtv y OBV con RBV durante 12 semanas alcanzaron RVS12 del 100%. Por otro lado, 96/99 (97%) de los pacientes con G1b y cirrosis compensada consiguieron una RVS12. Los resultados se incluyen en la tabla 7.
3. ESTUDIOS COMPLEMENTARIOS
3.1. PACIENTES TRATADOS CON AGONISTAS OPIÁCEOS
3.1.1. ESTUDIO M14-103
Estudio de fase II, multicéntrico, abierto, de un solo brazo, donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con RBV en adultos con HCC genotipo 1 que están recibiendo terapia con metadona o buprenorfina.
Tabla 7.- Resultados del estudio PEARL-I (M13-393).

OBV/PTV/rtv aumenta la concentración de buprenorfina en un 60% (40% con DSV). Su efecto sobre metadona es mínimo. Existían dudas sobre la influencia que metadona y buprenorfina pudieran tener en la exposición a los AAD, sobre todo en lo que respecta a OBV. Esos resultados dudosos se habían obtenido mediante comparaciones con controles históricos, en los que existía una gran dispersión en las exposiciones a OBV entre los diversos estudios. Dada esta limitación metodológica de los controles históricos, 38 pacientes no cirróticos G1 en tratamiento estable desde, al menos, 6 meses con metadona o buprenorfina ± naloxona fueron tratados durante 12 semanas con 3AAD + RBV. Los sujetos tratados tenían una edad promedio de 51 años (rango: entre 26 y 64); el 65,8% eran varones y el 5,3 % de raza negra. Una mayoría de los sujetos (86,8 %) tenía niveles basales en ARN VHC de, al menos, 800.000 UI/mL y una mayoría de ellos (84,2 %) tenía infección por genotipo 1a; el 68,4 % tenía el genotipo no-CC para la IL28B; el 15,8 % tenía una fibrosis portal (F2) y el 5,3 % tenía fibrosis avanzada (F3). El 94,7 % eran naïve al tratamiento para el VHC. La RVS12 fue 97% (37/38) lo que indica que la administración de los opiáceos no influye en la eficacia del tratamiento. Ningún sujeto experimentó fracaso virológico al tratamiento o recidiva.
4. POBLACIONES ESPECIALES
4.1. TRASPLANTE HEPÁTICO
4.1.1. ESTUDIO CORAL-I (M12-999)
Se dispone de los resultados de la cohorte 1 de este estudio fase II actualmente en curso, y que incluye 34 pacientes G1 trasplantados hepáticos con fibrosis estadio ≤2 (56% F0-F1; 44% F2). El estudio incluía un 85% de G1a, 79% de varones, edad media 60 años, 39,5 meses de media desde el trasplante y aclaramiento de creatinina medio de 90,5 mL/min. Los pacientes no habían recibido tratamiento postrasplante frente al virus C pero sí podían haberlo recibido previamente (IFN, pegilado o no, con o sin RBV). La pauta de tratamiento fue, en todos los casos, 3AAD + RBV 24 semanas. La dosis inicial de RBV, así como sus posibles modificaciones durante el período de tratamiento, se decidieron según criterio del investigador (dosis basal 600-800 mg/d en 56%; dosis al final del tratamiento de 600-800 mg/d en 68%; modificación de dosis en 56%). El ajuste de dosis de tacrolimus y ciclosporina estaba preestablecido.
Un total de 33/34 pacientes lograron RVS24 (97%): 5/5 en G1b, 28/29 en G1a. El paciente G1a no respondedor presentó una recaída post-tratamiento y había sido previamente tratado con PEG y RBV, con respuesta nula.
4.2. PACIENTES COINFECTADOS VIH/VHC)
4.2.1. ESTUDIO TURQUOISE-I (M14-004)
Estudio de fase II, multicéntrico, abierto, aleatorizado, donde se evalúa la eficacia y seguridad de la combinación OBV/PTV/rtv y DSV con RBV en adultos con HCC genotipo 1 coinfectados con VIH.
En la parte 1 (fase II) de este estudio en curso, 63 pacientes con G1 (89% G1a) coinfectados con VIH, fueron tratados con 3AAD + RBV durante 12 ó 24 semanas (1:1). Los principales datos demográficos eran: 92% hombres, edad media 50,9 años, 23% cirróticos (puntuación Child-Pugh ≤6), 67% naïve. Los regímenes antirretrovirales estaban basados en atazanavir y raltegravir. La cifra media de CD4 era 629 células/mL. La RVS12 fue del 93,5% (29/31) y 90,6% (29/32) en los grupos de 12 y 24 semanas, respectivamente. Por subgenotipos, las curaciones fueron 100% (7/7) en G1b y 91,1% (51/56) en G1a. Las causas de no respuesta en el brazo de 12 semanas fueron retirada del consentimiento en la semana 10 de tratamiento (ARN VHC indetectable en ese momento) y recaída post-tratamiento; en el brazo de 24 semanas las causas fueron fallo virológico durante el tratamiento en 1 paciente y reinfección post- tratamiento en 2 (tras estudio filogenético).
NOTA: Existen varios estudios en curso que vienen a completar la información en diversos subgrupos de pacientes como pacientes trasplantados con fibrosis avanzada, o bien sobre nuevos regímenes de tratamiento, aunque por el momento no se dispone de datos definitivos.
Seguridad
La seguridad de OBV, PTV y DSV se ha analizado según los resultados de 17 estudios fase I en voluntarios sanos que recibieron múltiples dosis de OBV, PTV y/o DSV, más los estudios en fase II y III hasta ahora revisados. En total, la base de datos de seguridad incluye alrededor de 4600 pacientes; el 95% de ellos recibieron más de 60 días de tratamiento. Los estudios SAPPHIRE I y II se realizaron frente a placebo. El 60% de los pacientes son ≤F2; no obstante, disponemos de resultados en fase III del estudio TURQUOISE-II, específicamente realizado en 380 pacientes con cirrosis compensada. RBV posee unos efectos secundarios bien tipificados tras años de uso. La incidencia de los efectos secundarios se resume en la Tabla 8.
La frecuencia de acontecimientos adversos fue similar en los pacientes tratados con placebo y en los tratados con OBV/PTV/rtv y DSV sin RBV. Los acontecimientos adversos graves fueron ligeramente superiores con 3AAD que con placebo aunque, en cualquier caso, muy escasos (1,2% y 0,4%, respectivamente). La frecuencia de acontecimientos adversos fue mayor en los pacientes tratados con el régimen triple incluyendo RBV, como se esperaba y son aquellos previamente descritos para RBV. La modificación de las dosis de RBV no influyó en la RVS. En el 8,5 % de sujetos que sí precisaron ajustes de la dosis de RBV durante el tratamiento, la tasa de RVS (98,5 %) fue equiparable a la conseguida por los sujetos que mantuvieron la dosis inicial de RBV durante todo el tratamiento.
La única muerte registrada en los ensayos clínicos aconteció en una paciente diabética de 64 años con cirrosis compensada que sufrió un shock circulatorio en el 9º día de tratamiento con AAD, el cual se atribuyó a acidosis láctica por metformina, con un fallo hepático en el contexto de un fracaso multiorgánico.
Los acontecimientos adversos más frecuentes con 3AAD + RBV fueron fatiga, astenia, cefalea, náusea, diarrea, prurito y exantema.
Las diferencias de riesgo indican que OBV/PTV/rtv y DSV pueden causar fatiga y prurito, el cual se ha observado con otros inhibidores NS3/4A y pudiera deberse a la influencia en transportadores biliares como BSEP.
Tabla 8.- Frecuencia de efectos secundarios.
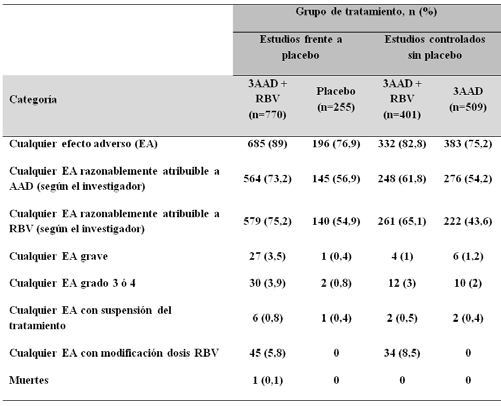
Once pacientes necesitaron ingreso hospitalario por acontecimientos adversos graves relacionados con el tratamiento (a juicio del investigador) que llevaron a la suspensión del mismo (Tabla 8). Las causas fueron: accidente cerebrovascular, hipoxia, dolor abdominal, artritis, pancreatitis, angioedema, hepatitis aguda, anemia, EPOC, acidosis láctica y celulitis.
En cuanto a las alteraciones en los parámetros analíticos, la anemia se produjo de acuerdo al uso o no de RBV, aunque puede que el DSV tenga un pequeño efecto en este sentido.
En los pacientes tratados con 3AAD sin RBV se recogieron un 6,7% (34/509) de anemias, todas ellas grado 1 (Hb>10 g/dL). En 1166 pacientes tratados con 3AAD + RBV hubo 653 casos de anemia (56%), entendida ésta como cualquier valor por debajo del límite inferior de normalidad. El 89,7% fueron grado 1 y el 9,8% grado 2. Hubo tres casos (0,4%) grado 3 (Hb 8-6,5 g/dL) y ninguno grado 4. Un total de 79/1.166 pacientes (6,7%) requirieron modificaciones de dosis, una suspensión de RBV (0,08%) y cinco tuvieron sendas necesidades de eritropoyetina o transfusión (0,2%). La hemólisis asociada a RBV se reflejó en un 12% de pacientes con bilirrubina >2xLSN (2% en pacientes sin RBV).
La mayoría de los pacientes presentaron una rápida normalización con el tratamiento en la cifra de transaminasas (GPT media -40 UI/L en 1ª semana de tratamiento). No obstante, en los estudios frente a placebo se apreció una elevación en GPT de, al menos, grado 2 (1,25-5xLSN) en el 2,2% de los pacientes (17/765) y de, al menos, grado 3 en el 1,2% (9/765) frente a un 16% y 3,9%, respectivamente, para los pacientes en los grupos placebo (atribuible a la fluctuación de las transaminasas por la propia hepatitis C). La adición de RBV no incrementó la incidencia de elevación de transaminasas en sangre (elevación grado 2 en GPT del 2% con 3AAD + RBV y del 1,8% con 3AAD sin RBV). De la suma de resultados en 2.626 pacientes con 3AAD ± RBV, 6 (0,2%) presentaron elevaciones de GPT grado 4 (>10xLSN). El principal responsable de la elevación de transaminasas es PTV, con un efecto dosis-dependiente. También presenta un riesgo bajo (3,8%) de hiperbilirrubinemia indirecta, aumentando si se asocia con RBV por la hemólisis inducida por ésta. Estas alteraciones hepáticas son transitorias, alcanzando el pico máximo en la semana 2, con posterior normalización pese a mantenimiento del tratamiento y sólo un 0,07% de los pacientes requirieron su suspensión por este motivo.
El análisis de regresión logística identificó el uso de estrógenos sistémicos concomitantemente con 3AAD como el principal factor de riesgo para la elevación de GPT, sobre todo grado 3. Este efecto no se identificó para progestágenos ni para estrógenos tópicos vaginales. La cirrosis tampoco fue un factor de riesgo. En un análisis posterior, se identificó a etinilestradiol (frente a estradiol, estriol y estrógenos conjugados) como el principal responsable. Con los datos actuales no hay evidencia de que PTV o la combinación 3AAD ± RBV sea causa de daño hepático severo con fallo hepático, incluyendo una relativamente larga serie de cirróticos. Ante el carácter infrecuente, leve y transitorio de la elevación de transaminasas no se recomienda su monitorización durante el tratamiento.
El OBV parece carecer de acontecimientos adversos específicos. DSV parece tener un mínimo efecto anemizante y una limitada capacidad dosis-dependiente para prolongar el intervalo QT, clínicamente no relevante a las dosis utilizadas.
El PTV es un inhibidor de OATP1B1 y los 3AAD de UGT1A1. Por lo tanto, esta combinación puede producir elevaciones de bilirrubina, predominantemente indirecta. Su incidencia es de un 3,8% (1,9% de ictericia) y su cénit se alcanza en la 1ª semana de tratamiento, seguido de un rápido descenso pese a mantenimiento del tratamiento. Un 0,2% de los pacientes presentaron complicaciones biliares (cólico biliar, colecistitis) durante el período de tratamiento.
No se han detectado diferencias de toxicidad por edad, sexo o grupo étnico. Los acontecimientos adversos más frecuentes en cirróticos compensados fueron similares a los de pacientes no cirróticos, aunque sí presentaron tendencia a una mayor frecuencia de hiperbilirrubinemia y anemia.
En trasplantados hubo una mayor incidencia de anemia (20%) que en no trasplantados. La modificación en la dosis de RBV fue necesaria en el 56%; 5 pacientes recibieron eritropoyetina; ninguno necesitó transfusión. No hubo en esta población mayor incidencia de hepatotoxicidad ni episodios de rechazo del injerto.
Los acontecimientos adversos en coinfectados con VIH fueron similares a los de monoinfectados.
En el momento actual, no existen datos de seguridad en pacientes cirróticos descompensados, pacientes con nefropatía crónica, mujeres embarazadas, niños ni con su uso en asociación con otros AAD.
INTERACCIONES MEDICAMENTOSAS
El perfil de interacciones de OBV/PTV/rtv y DSV es amplio, sobre todo porque ritonavir afecta a multitud de enzimas y transportadores. Se recomienda ver la ficha técnica para una descripción detallada de las mismas. Como norma general se desaconseja el uso de OBV/PTV/rtv y DSV, con o sin RBV, concomitantemente con inductores de CYP3A y CYP2C8, sustratos de CYP3A e inhibidores de CYP3A4 y CYP2C8. Su administración con etinilestradiol supone un aumento del riesgo de hepatotoxicidad. En pacientes trasplantados es relevante resaltar que el uso concomitante de estos AAD con ciclosporina o tacrolimus afecta sus niveles plasmáticos y requiere un ajuste de dosis y una monitorización estrecha.
DISCUSIÓN
De forma general, es importante destacar que el programa de desarrollo de OBV/PTV/rtv y DSV dispone de un importante número de ensayos clínicos que incluyen un gran número de pacientes, lo que permite sacar conclusiones sólidas en las poblaciones estudiadas. Asimismo, incluye ensayos clínicos con placebo como comparador, lo que permite una evaluación adecuada del perfil de seguridad de OBV/PTV/rtv y DSV.
Sin embargo, todos los estudios comparan la eficacia del tratamiento con la eficacia de controles históricos (telaprevir asociado a interferón y ribavirina). La validez de esta comparación se desconoce ya que no está claro si las características de los pacientes del brazo de tratamiento eran similares a los pacientes evaluados en el control histórico (potencial sesgo de confusión). Tampoco está claro si los pacientes de ambos grupos de tratamiento se trataron de igual forma en relación al tratamiento médico base recibido. Se empleó un margen de no inferioridad de 10,5%, valor que no se justificó.
No existen estudios de comparación directa entre OBV/PTV/rtv y DSV frente a otras alternativas sin interferón, Sofosbuvir + Daclatasvir, Sofosbuvir/Ledipasvir o Sofosbuvir + Simeprevir.. Sin embargo, dado que los diferentes ensayos emplearon la misma variable principal de estudio y las poblaciones son comparables, aunque no sea posible establecer una comparación cuantitativa con resultados y validez estadística, si nos permiten contextualizar los resultados obtenidos en función de las tasas de respuesta obtenidas.
A continuación se resaltan algunos aspectos importantes a considerar sobre el tratamiento con la combinación de OBV/PTV/rtv y DSV.
GENOTIPO 1
En los estudios fase III se demuestra una eficacia superior al 95% cuando los pacientes G1b no cirróticos son tratados con OBV/PTV/rtv y DSV, independientemente de una posible no respuesta a tratamiento previo con PEG/RBV u otros factores del paciente (Tabla 9). Por ello, de forma general, el régimen recomendado en pacientes con genotipo 1b no cirróticos debe ser OBV/PTV/rtv y DSV 12 semanas.
Tabla 9.- Resumen de resultados en fase III: G1b.
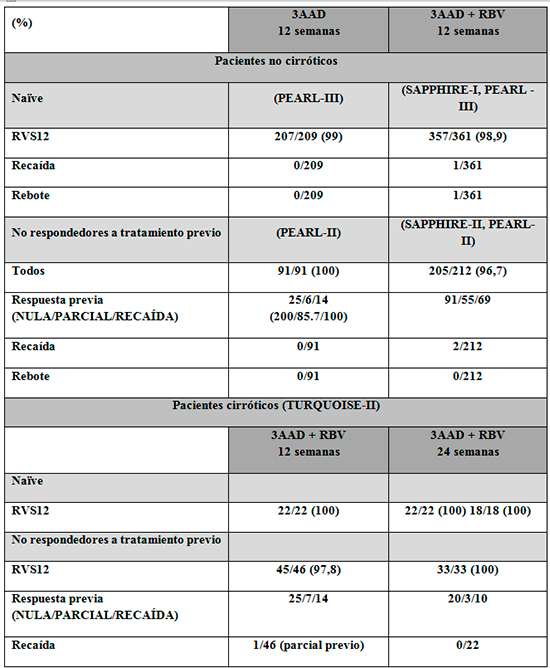
Teniendo en cuenta los resultados del estudio AVIATOR, probablemente la combinación OBV/PTV/rtv + RBV proporcione similares resultados a OBV/PTV/rtv y DSV, habiéndose optado por ésta última ante el mejor perfil de seguridad de DSV frente al de RBV. No existen datos en no respondedores previos a otros AAD.
En cuanto al G1a (Tabla 10), en el estudio PEARL-IV la combinación OBV/PTV/rtv y DSV sin RBV no demostró la no inferioridad frente a la misma pauta con RBV, ambas durante 12 semanas y en pacientes naïve (RVS 97% con RBV, 90,2% sin RBV). Basándose en todo ello, la recomendación para G1a no cirróticos es la combinación OBV/PTV/rtv y DSV + RBV, tanto en pacientes naïve como en pretratados, durante 12 semanas.
Tabla 10.- Resumen de resultados en fase III: G1a.
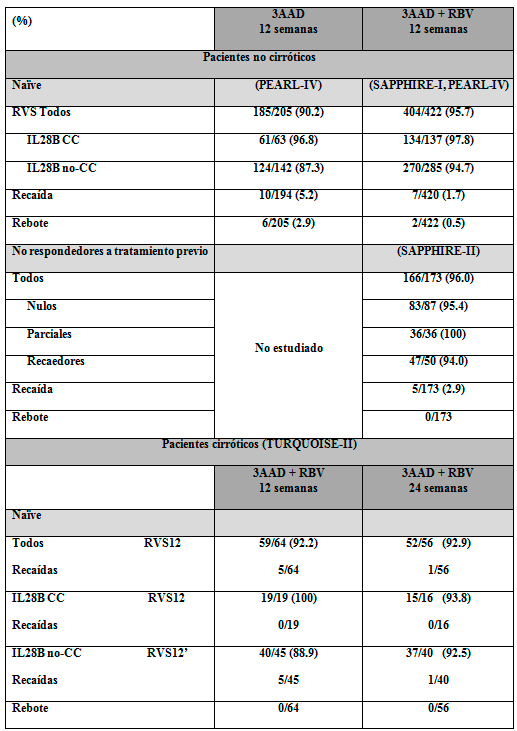

En general, en pacientes cirróticos la RVS tiende a ser inferior. En el estudio TURQUOISE-II la combinación OBV/PTV/rtv y DSV + RBV se comparó durante 12 ó 24 semanas en pacientes cirróticos G1, previamente tratados (con PEG/ RBV) o naïve. Los resultados en pacientes G1b, tratados durante 12 semanas (RVS 98,5%), muestran tasas de respuestas similares a los tratados durante 24 semanas, por tanto en pacientes genotipo 1b cirróticos el régimen recomendado es OBV/PTV/rtv y DSV con RBV durante 12 semanas.
En pacientes infectados con VHC G1a cirróticos naïve, la RVS fue ligeramente inferior a G1b con un 92-95%, no existiendo diferencias entre 12 y 24 semanas de tratamiento; en los pacientes previamente tratados, y a pesar del bajo número de pacientes en cada subgrupo, parece que tampoco hay diferencias entre 12 y 24 semanas de tratamiento en pacientes con respuesta parcial previa a biterapia o recidiva. Sin embargo, en pacientes con genotipo 1a, la respuesta nula previa sí que se ha asociado a una menor probabilidad de alcanzar RVS. Por ello, en estos y en aquellos con valores de laboratorio (AFP, plaquetas y albúmina) desfavorables, indicativos de mayor riesgo de recaída, debería considerarse la ampliación a 24 semanas.
GENOTIPOS 2 Y 3
OBV/PTV/rtv y DSV no son eficaces o no han sido suficientemente estudiados en genotipos 2 y 3.
GENOTIPO 4
En pacientes G4 no cirróticos la combinación OBV/PTV/rtv + RBV obtuvo en el estudio PEARL-1 un 100% de RVS, tanto en pacientes naïve como en no respondedores a tratamiento previo. Por el momento no se disponen de datos en pacientes cirróticos compensados con genotipo 4, aunque actualmente se encuentra en marcha el ensayo M11-665 en el cual se estudia el tratamiento de pacientes cirróticos compensados G4 con OBV/PTV/rtv + RBV, en pautas de 12, 16 y 24 semanas. La actividad in vitro de PTV y OBV es similar para G1b y G4 y ambos medicamentos presentan una barrera genética similar frente a la aparición de resistencias para ambos genotipos. Por tanto, hasta que los resultados del estudio M11-665 estén disponibles, la pauta en cirróticos compensados G4 debe ser 24 semanas.
OBV/PTV/ RTV Y DSV EN POBLACIONES ESPECIALES
El análisis intermedio del estudio CORAL-I indica que la combinación de los 3 AAD + RBV durante 24 semanas proporciona tasas de RVS muy elevadas (97%) en pacientes trasplantados hepáticos G1 sin fibrosis avanzada. Sin embargo, estos resultados proceden de 33/34 pacientes con grado de fibrosis F2 o inferior y naïve al tratamiento post-trasplante, tratados con dosis variables de RBV en función del criterio del investigador, por lo que habrá que esperar a la publicación de los resultados definitivos para establecer la mejor pauta de tratamiento en estos pacientes. Este estudio no incluye a pacientes con cirrosis avanzada ni con una enfermedad recurrente agresiva.
Estas tasas son similares a las alcanzadas con otros regímenes sin interferón en los subgrupos de pacientes con enfermedad recurrente post-trasplante menos avanzada. En los pacientes con hepatitis C recurrente post-TH del estudio SOLAR-1, las tasas de RVS12 con SOF/LDV + RBV fueron muy elevadas en pacientes con fibrosis F0-F3 (96-98%) y en aquellos con cirrosis compensada (Child Pugh A) del 96%. La tasa de RVS12 en pacientes con cirrosis descompensada fue menor (Child Pugh B: 83-85%; Child Pugh C: 60-67%), aunque los porcentajes en los pacientes con cirrosis en estadio Child-Pugh C deben interpretarse con precaución debido al escaso número de pacientes en cada brazo de tratamiento. En general, la prolongación del tratamiento a 24 semanas no supone un incremento en las tasas de RVS12, aunque el número de pacientes en cada subgrupo es pequeño (19 y 20). En un estudio reciente de práctica real, con las limitaciones metodológicas que los resultados conllevan con respecto a los obtenidos en un ensayo clínico, la combinación SMV + SOF ± RBV durante 12 semanas consiguió una tasa de RVS12 del 90% en pacientes con genotipo 1, 30% de los cuales tenían enfermedad avanzada (F3-F4) y 11% recidiva colestásica. En pacientes con genotipo 1a, la tasa de RVS12 fue significativamente inferior en los pacientes con F3-F4 (71%) frente a aquellos con F0-F2 (91%), no existiendo esas diferencias en los pacientes con genotipo 1b (21). Datos preliminares de los estudios de vida real de la combinación DCV+SOF ± RBV x24 semanas en pacientes con recurrencia post-trasplante grave proporcionan tasas de RVS12 aproximadamente del 75% (22). Datos de la cohorte CUPILT de 30 pacientes con recurrencia grave (hepatitis colestásica fibrosante) tratados con DCV+SOF durante 24 semanas muestran tasas RVS12 en los pacientes que ya han finalizado el seguimiento a las doce semanas del 100% (11/11) (23). SOF/LDV tiene, al igual que DCV + SOF, la ventaja de que puede utilizarse en pacientes con cirrosis descompensada (Child-Pugh B y Child-Pugh C) -y en tener actividad frente al genotipo 3. Mientras que simeprevir requiere especial precaución si se administra en pacientes con insuficiencia hepática moderada o grave (Child-Pugh B o C) y OBV/PTV/rtv y DSV están contraindicados en insuficiencia hepática grave (Child- Pugh C).
Si bien no se dispone de datos en poblaciones especiales con otros genotipos, los resultados de los estudios de fase III permitirían la extrapolación de los resultados del estudio SOLAR a otros genotipos.En pacientes coinfectados con VIH se reproducen los resultados de curación de los pacientes monoinfectados, tanto en duración de la terapia como por subgenotipos 1.
SEGURIDAD
En general, OBV/PTV/rtv y DSV presentan un buen perfil de seguridad y una buena tolerancia, con interrupciones excepcionales del tratamiento debidas a acontecimientos adversos y una incidencia baja de acontecimientos adversos graves.
La complejidad de las interacciones hace altamente recomendable, antes de iniciar del tratamiento o ante la posible necesidad de otros medicamentos concomitantes durante el mismo, la consulta de la ficha técnica así como de bases de datos actualizadas donde esté ampliamente descrito el perfil de interacciones farmacológicas.. Esto resulta de especial importancia en el manejo de pacientes trasplantados con tratamiento inmunosupresor.
CONCLUSIÓN
La combinación de OBV/PTV/rtv y DSV es el primer régimen que se autoriza sin interferón que combina antivirales de acción directa y que no incluye a sofosbuvir como parte del régimen.
El régimen OBV/PTV/rtv y DSV con RBV durante 12 semanas es una alternativa terapéutica a otras combinaciones de AAD, en pacientes con genotipo 1, tanto mono como coinfectados por VIH, con y sin cirrosis. En pacientes con genotipo 1b no cirróticos se puede prescindir de la RBV. En pacientes con genotipo 1a y cirrosis compensada, con respuesta nula a tratamiento previo con PEG/RBV o factores predictores de mala respuesta, se recomienda prolongar el tratamiento a 24 semanas.
En pacientes con genotipo 4, el régimen OBV/PTV/rtv con RBV administrado durante 12 semanas es una alternativa terapéutica a otras combinaciones de AAD. Sin embargo, en pacientes cirróticos la evidencia procede de la extrapolación de resultados en pacientes con genotipo 1b y la duración óptima del tratamiento es desconocida. En estos casos, se recomienda emplear LDV/SOF (evidencia todavía escasa) o SMV/SOF, ambos durante 12 semanas.
Según los datos preliminares del estudio CORAL-I, OBV/PTV/rtv y DSV + RBV durante 24 semanas es una alternativa terapéutica eficaz y segura en pacientes con hepatitis C genotipo 1 trasplantados hepáticos, en los que la enfermedad recurre de una forma no agresiva y el grado de fibrosis es leve. Se debe de tener en cuenta el perfil de interacciones de esta combinación, que complica el manejo de la inmunosupresión. En el contexto del post-trasplante, en el momento actual existen otras combinaciones (SOF/LDV + RBV o SOF/SMV + RBV) que con tratamientos de 12 semanas obtienen resultados similares. En los pacientes con fibrosis avanzada y/o descompensados, la ausencia de datos con esta combinación hace preferible el uso de otras alternativas terapéuticas.
No se disponen de datos del uso de esta combinación en pacientes con VHC G1 con fracaso al tratamiento con inhibidores de la proteasa ni en pacientes con cirrosis descompensada. Debido a la presencia del IP (PTV) el uso en pacientes con Child-Pugh C está contraindicado.
No hay datos en niños, pacientes con insuficiencia renal y trasplantados hepáticos con fibrosis avanzada.
El uso de OBV/PTV/rtv y DSV no está indicado en pacientes con infección por los genotipos 2 y 3.
Tanto OBV/PTV/rtv como DSV presentan interacciones con otros medicamentos que obligan a su valoración específica.
REFERENCIAS
1. Kowdley KV, Lawitz E, Poordad F, et al. Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med. 2014 Jan 16;370(3):222-32
2. Kwo PY, Mantry PS, Coakley E, el al. An interferon-free antiviral regimen for HCV after liver transplantation. N Engl J Med. 2014 Dec 18;371(25):2375-82.
3. Eron JJ, Lalezari J, Slim J, et al. Safety and efficacy of ombitasvir - 450/r and dasabuvir and ribavirin in HCV/HIV-1 co-infected patients receiving atazanavir or raltegravir ART regimens. J Int AIDS Soc. 2014 Nov 2;17(4 Suppl 3):19500
4. Poordad F, Agarwal K, Younes Z, Cohen D, Xie W, Podsadecki T. Low Relapse Rate Leads to High Concordance of Sustained Virologic Rlesponse (SVR) at 12 Weeks With SVR at 24 Weeks After Treatment With ABT-450/Ritonavir, Ombitasvir, and Dasabuvir Plus Ribavirin in Subjects With Chronic Hepatitis C Virus Genotype 1 Infection in the AVIATOR Study. Clin Infect Dis. 2015 Feb 15;60(4):608-10.
5. Lawitz E, Sullivan G, Rodriguez-Torres M, et al. Exploratory trial of ombitasvir and ABT-450/r with or without ribavirin for HCV genotype 1, 2, and 3 infection. J Infect. 2015 Feb;70(2):197-205.
6. Andreone P, Colombo MG, Enejosa JV, et al. ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology. 2014 Aug;147(2):359-365.
7. Ferenci P, Bernstein D, Lalezari J, et al. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014 May 22;370(21):1983-92.
8. Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014 May 22;370(21):1973-82.
9. Feld JJ, Kowdley KV, Coakley E, et al. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014 Apr 24;370(17):1594-603.
10. Zeuzem S, Jacobson IM, Baykal T, et al. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014 Apr 24;370(17):1604-14.
11. Informe de Posicionamiento Terapéutico de Sofosbuvir (Sovaldi®). Disponible en: http://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-sofosbuvir-sovaldi.pdf
12. Informe de Posicionamiento Terapéutico de Daclatasvir (Daklinza®). Disponible en: http://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-daclatasvir-daklinza.pdf
13. Informe de Posicionamiento Terapéutico de Simeprevir (Olysio®). Disponible en: http://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-simeprevir-olysio.pdf
14. Informe de Posicionamiento Terapéutico de Sofosbuvir/Ledipasvir (Harvoni®). Pendiente de publicación.
15. EPAR de Exviera®. Disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003837/WC500182235.pdf
16. EPAR de Viekirax®. Disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003839/WC500183999.pdf .
17. Ficha Técnica de Exviera® disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/003837/WC500182233.pdf
18. Ficha Técnica de Viekirax®. Disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003839/WC500183997.pdf
19. Flamm SL, Everson GT, Charlton M, et al. LDV/SOF +RBV for the Treatment of HCV in Patients with Decompensated Cirrhosis: Preliminary Results of a Prospective, Multicenter Study. Oral communication #239 presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA.
20. Reddy R. LDV/SOF + RBV for the Treatment of HCV in Patients with Post Transplant Recurrence: Preliminary Results of a Prospective, Multicenter Study. Oral communication #8 presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA.
21. Pungpapong S, et al. Multicenter experience using sofosbuvir and simeprevir with/without ribavirin to treat HCVgenotype 1 after liver transplantation. American Association for the Study of Liver Diseases (AASLD) Liver Meeting. Boston, November 7- 12, 2014; Abstract 9.
22. Fontana R, Bahirwani R, Reddy R, et al. High Efficacy and Favorable Safety Profile of Daclatasvir Based All Oral Antiviral Therapy in Liver Transplant Recipients with Severe Recurrent HCV. American Association for the Study of Liver Diseases (AASLD) Liver Meeting. Boston, November 7-12, 2014. AbstractLB-22.
23. Leroy V, Dumortier J, Coilly A, et al. High Rates Of Virological Response And Major Clinical Improvement during Sofosbuvir and Daclatasvir-based Regimens for the Treatment of Fibrosing Cholestatic HCV-Recurrence after Liver Transplantation: The ANRS CO23 CUPILT Study. American Association for the Study of Liver Diseases (AASLD) Liver Meeting. Boston, November 7-12, 2014. Abstract21
GRUPO DE EXPERTOS
(por orden alfabético)
Agencia Española de Medicamentos y Productos Sanitarios
Centro de Información Farmacoterapéutica del Servizo Galego de Saúde
Comunidad Autónoma de Andalucía
Comunidad Autónoma de Castilla- La Mancha
Grupo Técnico de Utilidad de Medicamentos de la Comunidad de Madrid
Antonio Olveira
Servicio de Aparato Digestivo (Unidad de Hepatología). Hospital Universitario La Paz, Madrid
Arantxa Sancho López
Servicio de Farmacología Clínica. Hospital Universitario Puerta de Hierro Majadahonda, Madrid. Miembro del Comité de Medicamentos de Uso Humano (CHMP) de la EMA.
Todos los expertos han realizado una declaración de conflictos de interés.
El Laboratorio Titular, la Sociedad Española de Trasplante Hepático, Sociedad Española de Farmacología Clínica, Sociedad Española de Farmacia Hospitalaria, Asociación Española para el Estudio del Hígado, Federación Nacional de Enfermos y Trasplantados Hepáticos, la Federación Española de Hemofilia y el Foro Español de Activistas en Tratamientos del VIH han tenido oportunidad de enviar comentarios al documento, si bien el texto final es el adoptado por el GCPT.
Fe de erratas
Con fecha 14 de abril de 2015, se ha corregido la siguiente errata:
En el Grupo de Expertos, se ha eliminado al “Comité de Evaluación de Nuevos Medicamentos en el Ámbito Hospitalario de Euskadi” y se ha incluido a la “Comunidad Autónoma de Castilla- La Mancha”.
Fuente: Agencia Española de Medicamentos y Productos Sanitarios.
