Informe de Posicionamiento Terapéutico de simeprevir (Olysio®)
INFORME DE POSICIONAMIENTO TERAPÉUTICO PT-SIMEPREVIR/V1/01112014
Informe de Posicionamiento
Terapéutico de Simeprevir (Olysio®)
Fecha de publicación: 18 de noviembre de 2014
Fecha de corrección: 20 de noviembre de 2014 (ver al final)
La infección por el virus de la hepatitis C (VHC) es un problema de salud de primera magnitud en Europa y especialmente en los países mediterráneos, donde la tasas de prevalencia oscilan entre el 1-3%. Es la primera causa de enfermedad hepática terminal y una de las principales indicaciones de trasplante hepático. La recurrencia de la infección en el órgano trasplantado y un curso más agresivo y acelerado hacen que los resultados a medio plazo del trasplante hepático sean peores que los observados en cirrosis de otras etiologías (1).
El VHC se divide en 7 genotipos con numerosos subtipos, siendo el genotipo 1 (principalmente 1b) el más frecuente en Europa con una prevalencia aproximada del 70%, seguido del genotipo 3. En España, el genotipo 4 es el tercero en frecuencia, siendo el genotipo 2 el más infrecuente. El genotipo del VHC, si bien no condiciona una evolución clínica diferente de la hepatitis C crónica (HCC), sí tiene un gran impacto en la respuesta al tratamiento (1).
En el momento actual, los tratamientos aprobados para la HCC son interferón pegilado (PEG), ribavirina (RBV), dos inhibidores de la proteasa (IP) NS3/4A, boceprevir (BOC) y telaprevir (TVR), activos sólo frente al genotipo 1 y Sofosbuvir (SOF), un inhibidor nucleósido de la polimerasa NS5B con actividad frente a todos los genotipos del VHC. La terapia triple con PEG/RBV y alguno de los inhibidores de proteasa están disponibles para las infecciones por el genotipo 1 desde el año 2011 y presentan una tasa de respuesta viral sostenida (RVS) que oscila entre el 30-80% en función de subtipo, el genotipo de la IL28B, la respuesta al tratamiento previo y la gravedad del daño hepático. Para el resto de los genotipos, el tratamiento consiste en la combinación de PEG/RBV durante 16-48 semanas. Las tasas de RVS oscilan entre el 30 y el 80%, siendo en los genotipos 2 y 3 en los que se consiguen mejores resultados. En pacientes naïve con genotipo 1 y 4, las terapias basadas en la combinación de PEG + RBV con SOF incrementan de forma significativa las tasas de RVS (2).
Todos estos tratamientos basados en interferón se asocian a un gran número de efectos adversos, especialmente en los pacientes con hepatopatías más avanzadas, lo que limita su aplicabilidad en algunos grupos de pacientes. Estos efectos adversos incluyen riesgo de descompensación hepática, sepsis e importante mielosupresión. Los efectos adversos psiquiátricos de tipo ansioso-depresivo y el insomnio también son muy frecuentes (3,4)
Por lo tanto, las regulares tasas de eficacia y la gran cantidad de efectos secundarios de las terapias basadas en interferón, especialmente en los pacientes con enfermedad hepática más avanzada y en los que han fracasado tratamientos previos, hace muy necesarios otros fármacos que sean mejor tolerados, más efectivos y que permitan realizar tratamientos libres de PEG (5-10).
En el momento actual existe un enorme desarrollo de la investigación de nuevos fármacos frente al VHC que actúan de manera directa y pertenecen a 4 clases diferentes: los inhibidores de proteasa NS3/4A, los inhibidores de NS5A y los inhibidores de la polimerasa NS5B que pueden ser nucleósidos o nucleótidos. Todas estos nuevas moléculas se están estudiando en diversas combinaciones (incluyendo o no PEG y/o RBV) entre agentes de las diferentes clases, observándose aumentos muy importantes en la eficacia (2).
SIMEPREVIR (OLYSIO®)
Simeprevir (SMV) ha sido autorizado para el tratamiento de la HCC en adultos en combinación con otros fármacos activos frente al VHC de acuerdo a ficha técnica. SMV se presenta en cápsulas duras y contiene 150 mg de principio activo. La dosis recomendada es de 150 mg (1 cápsula) al día durante 12 semanas y debe tomarse con alimentos.
Farmacología
SMV es un inhibidor específico de la proteasa NS3/4A, esencial para la replicación viral, utilizado en combinación con otros fármacos activos frente al VHC en función del genotipo.
SMV es activo frente a los genotipos 1,4, 5 y 6. Su eficacia en pacientes con VHC genotipos 5 y 6 no ha sido estudiada en ensayos clínicos, por lo que no se debe utilizar en estos pacientes. En el genotipo 1a, la presencia del polimorfismo Q80K afecta negativamente a la respuesta virológica de SMV. Se dispone de un total de 27 estudios del desarrollo farmacológico clínico de SMV, 4 estudios en la fase 2 II y 6 estudios en la fase 3 (11, 12).
Posología
Las Tablas 1 y 2 muestran los medicamentos concomitantes recomendados y la duración del tratamiento para la terapia combinada de simeprevir, y las reglas de parada, respectivamente.
Tabla 1
| Población de pacientes | Tratamiento | Duración |
| Pacientes no tratados previamente (naïve) y con recidiva previa con VHC genotipo 1 ó 41 | OLYSIO + peginterferón alfa + ribavirina2 | 24 semanas3 El tratamiento con OLYSIO se debe iniciar en combinación con peginterferón alfa y ribavirina y ser administrado durante 12 semanas y posteriormente continuar durante 12 semanas adicionales con peginterferón alfa y ribavirina. |
| Pacientes sin respuesta previa (incluidos los pacientes que hayan tenido una respuesta parcial o nula) con VHC genotipo 1 ó 41 | OLYSIO + peginterferón alfa + ribavirina2 | 48 semanas El tratamiento con OLYSIO se debe iniciar en combinación con peginterferón alfa y ribavirina y ser administrado durante 12 semanas y posteriormente continuar durante 36 semanas adicionales |
| Pacientes con VHC genotipo 1 ó 4, independientemente del historial de tratamiento previo4 | OLYSIO + sofosbuvir (+/- ribavirina)5 | 12 semanas (ver secciones 4.4, 4.8 y 5.1) |
1 Incluye a pacientes con o sin cirrosis y aquellos pacientes coinfectados con el virus de la inmunodeficiencia humana (VIH). Recidiva o sin respuesta a un tratamiento previo con interferón (pegilado o no pegilado), con o sin ribavirina (ver sección 5.1).
2Cuando se valore utilizar un tratamiento combinado de OLYSIO con peginterferón alfa y ribavirina en los pacientes con VHC genotipo 1a, antes de comenzar el tratamiento se debe realizar un test de identificación del polimorfismo Q80K en NS3 (ver sección 4.4).
3Los pacientes no tratados previamente (naïve) y con recidiva previa con cirrosis que están coinfectados con el VIH deben recibir 48 semanas de tratamiento. El tratamiento con OLYSIO se debe iniciar en combinación con peginterferón alfa y ribavirina y ser administrado durante 12 semanas y posteriormente continuar durante 36 semanas adicionales con peginterferón alfa y ribavirina. Ver sección Poblaciones especiales de pacientes - Coinfección por el VHC y el virus de la inmunodeficiencia humana de tipo 1 (VIH-1).
4Incluye a pacientes no tratados previamente (naïve) o pacientes que fracasaron a un tratamiento previo con peginterferón alfa y ribavirina con o sin cirrosis.
5El tratamiento de OLYSIO con sofosbuvir solamente se debe utilizar en pacientes que no toleran o no son candidatos al tratamiento con interferón, y tienen una necesidad de tratamiento urgente. Se puede añadir ribavirina en base a la evaluación clínica individual de cada paciente (ver secciones 4.4, 4.8 y 5.1). La duración del tratamiento recomendada es de 12 semanas. Se puede valorar una duración de tratamiento de OLYSIO con sofosbuvir (con o sin ribavirina) más larga (hasta 24 semanas) en base a las características individuales (ver secciones 4.4, 4.8 y 5.1).
| ARN del VHC | Medida |
| Semana 4 de tratamiento: ≥ 25 | Interrupción de simeprevir, peginterferón alfa y ribavirina |
| Semana 12 de tratamiento: detectable | Interrupción del peginterferón alfa y la ribavirina (el tratamiento con simeprevir está completo en la semana 12) |
| Semana 24 de tratamiento: detectable | Interrupción del peginterferón alfa y la ribavirina |
Tabla 2. Reglas de interrupción del tratamiento en los pacientes que reciben simeprevir en combinación con peginterferón alfa y ribavirina con respuesta virológica insuficiente durante el tratamiento
Eficacia
La eficacia de SMV ha sido evaluada en dos estudios de fase 3 junto a PEG y RBV en pacientes con VHC genotipo 1 no tratados previamente y en un estudio de fase 3 junto PEG y RBV en pacientes con VHC genotipo 1 tratados previamente.
El programa clínico incluye también 4 estudios de soporte en pacientes con VHC genotipo 4, co-infectados, regímenes libres de interferón y eficacia a largo plazo.
Genotipo 1 en pacientes sin tratamiento previo
Estudio C208 (QUEST-1)
Estudio de fase III multicéntrico, doble ciego, aleatorizado, controlado con placebo (PBO) para investigar la eficacia de SMV 150 mg/día durante 12 semanas en combinación con interferón pegilado alfa 2a 180 μ g subcutáneos/semana y RBV 1.000-1.200 mg/día durante 24 o 48 semanas en función de la respuesta al tratamiento (TGR).
Los pacientes se estratificaron en función del subtipo de genotipo 1 del VHC (1a, 1b, otros) y del polimorfismo del gen IL28B.
En las primeras 12 semanas, los pacientes debían recibir SMV o PBO en combinación con PEG/RBV, seguido de 12 semanas adicionales de PEG/RBV. Aquellos pacientes con ARN VHC <25 UI/mL (detectable o indetectable) en la semana 4 y <25 UI/mL e indetectable en la semana 12, finalizaban el tratamiento en la semana 24. El resto, debían continuar con PEG/RBV hasta la semana 48.
Se incluyeron pacientes que no habían recibido tratamiento previo con PEG + RBV (naïve) con infección activa VHC con genotipo 1, enfermedad hepática compensada, sin coinfección por el virus de la hepatitis B (VHB) o por el virus de la inmunodeficiencia humana (VIH).
El objetivo principal fue comparar la eficacia y seguridad del tratamiento con SMV + PEG + RBV con PEG alfa 2a + RBV, que era considerado el tratamiento estándar al inicio del estudio. La variable principal del estudio fue el porcentaje de pacientes con respuesta viral sostenida en la semana 12 postratamiento (RVS12).
Se incluyeron 394 pacientes con una edad media de 46 años, 56,3% varones, mayoritariamente de raza blanca. En el grupo placebo había menos pacientes de raza negra que en el grupo de SMV (3,1% vs 10,3%). El 56,1% tenían VHC genotipo 1a, 43,9% 1b. El polimorfismo Q80K estaba presente al inicio en el 23,3% del total de los pacientes incluidos en el estudio y en el 41,1% de los infectados con el genotipo 1a. De acuerdo a la escala METAVIR, el 43,1% de los pacientes tenían F0-F1, el 26,9% tenían F2, y el 17,7% F3. Los pacientes con cirrosis hepática compensada (F4) estuvieron escasamente representados (12,3%), así como los pacientes con genotipo favorable (CC) de IL28B (28,9%).
En el análisis por intención de tratar, SMV en combinación con PEG/RBV demostró superioridad a PBO en combinación con PEG/RBV. El 79,4% de los pacientes del brazo de SMV alcanzaron RVS12en comparación con el 50,1% en el brazo de PBO (Tabla 3). Los resultados de RVS24fueron similares (83% vs 60%).
Los datos de eficacia de SMV fueron consistentes en todos los subgrupos analizados (carga basal, sexo, polimorfismo IL28B, Metavir) a excepción del polimorfismo Q80K.
La RVS12 en los pacientes con genotipo 1a y polimorfismo Q80K presente tratados con SMV fue 51,7% frente al 53% del grupo PBO; mientras que la respuesta de los pacientes sin el polimorfismo fue 84,9% para SMV y 44% para PBO. En pacientes con genotipo 1a y polimorfismo Q80K, la diferencia entre SMV y placebo no fue estadísticamente significativa. La RVS12 en pacientes con VHC genotipo 1b tratados con SMV fue de 90% frente al 52% en el grupo PBO.
De acuerdo a los criterios de Tratamiento Guiado por Respuesta (TGR), 84,4% de los pacientes del brazo de SMV recibieron un total de 24 semanas de tratamiento con PEG/RBV, de los cuales 90,6% alcanzaron RVS12.
Estudio C216 (QUEST-2)
El diseño de este estudio es idéntico al anterior, la principal diferencia es que los pacientes podían recibir tratamiento con PEG alfa 2a o 2b.
Se incluyeron 391 pacientes con una edad media de 47 años, 55,5% varones, mayoritariamente de raza blanca. El 40,7% tenían genotipo 1a y 58,1% 1b. El polimorfismo Q80K estaba presente al inicio en el 10,2% de los pacientes del estudio (todos menos uno, infectados con VHC genotipo 1a) y en el 24,2% de los pacientes con genotipo 1a. De acuerdo a la escala METAVIR, el 49,7% de los pacientes tenían F0-F1, el 28% tenían F2, el 13,9% F3 y el 8,4% F4. Hubo más pacientes con cirrosis hepática compensada (F4) en el brazo PBO (11,2%).
En el análisis por intención de tratar, SMV en combinación con PEG/RBV fue superior a PBO en combinación con PEG/RBV. El 81,9% de los pacientes del brazo de SMV alcanzaron RVS12 en comparación con el 49,7% en el brazo de PBO (Tabla 3). Los resultados de RVS24 (81,4% vs 45,9%) fueron similares.
| QUEST-1 | QUEST-2 | QUEST-1/ QUEST-2 | ||||
| Naïve | ||||||
| SMV | PBO | SMV | PBO | SMV | PBO | |
| N=264 | N=130 | N=257 | N=134 | N=521 | N=264 | |
| RVS12 | 210/264 | 65/130 | 209/257 | 67/134 | 419/521 | 132/264 |
| % | 79,4% | 50,1% | 81,9% | 49,7% | 80,4% | 50% |
| Diferencia | 29,3% (IC 20,1-38,6) p<0,001 | 32,2% (IC 23,3-41,2) p<0,0001 | 30,5% (IC 24,1-36,9) p<0,001 | |||
| RVS12 cirrosis (F4) | 18/31 | 5/17 | 11/17 | 6/15 | 29/48 | 11/32 |
| % | 58% | 29,5% | 65% | 40% | 66,3% | 34,4% |
| Diferencia | 40,7% (IC 29,5-51,8) p<0,0001 | 47,5% (IC 35,2;59,9) p<0,0001 | 43,0% (IC 34,9-51,1) | |||
Tabla 3. Resumen de la variable principal (RVS12) de los estudios pivotales en pacientes naïve (Análisis ajustado por ITT)
Al igual que en el ensayo clínico anterior, los datos de eficacia de SMV fueron consistentes en todos los subgrupos analizados (subtipo, carga basal, sexo, polimorfismo IL28B, Metavir) a excepción del polimorfismo Q80K.
En el brazo de SMV, la RVS12 en los pacientes con genotipo 1a y polimorfismo Q80K presente tratados con SMV fue 75% frente a 50% en el grupo PBO y en los pacientes sin este polimorfismo, 82.3% para SMV y 43% en PBO. En este estudio, en los pacientes con genotipo 1a y polimorfismo Q80K, aunque la RVS de SMV fue numéricamente mayor que en el grupo PBO, esta diferencia no fue estadísticamente significativa. La RVS12 en pacientes con VHC genotipo 1b tratados con SMV fue de 82% frente al 53% en el grupo PBO.
De acuerdo a los criterios de TGR, 91.4% de los pacientes del brazo de SMV recibieron un total de 24 semanas de tratamiento con PEG/RBV, de los cuales 86% alcanzaron RVS12.
Genotipo 1 en pacientes pretratados
Estudio HPC3007 (PROMISE)
Estudio de fase 3 multicéntrico, aleatorizado (2:1), controlado con placebo, doble ciego, para evaluar la eficacia y seguridad de SMV 150 mg/día durante 12 semanas en combinación con interferón pegilado alfa 2a 180 μ g subcutáneos/semana y RBV 1.000-1.200 mg/día durante 24 o 48 semanas en función de la respuesta al tratamiento.
Se incluyeron pacientes con infección activa por VHC con genotipo 1 que habían recaído después de un tratamiento previo basado en interferón. Los pacientes debían carecer de antecedentes de descompensación hepática y no estar coinfectados por VHB o VIH.
El objetivo principal fue comparar la eficacia y seguridad del tratamiento con SMV + PEG + RBV frente a PEG + RBV. La variable principal del estudio fue el porcentaje de pacientes con respuesta viral sostenida en la semana 12 postratamiento (RVS12).
Se incluyeron 393 pacientes con una edad media de 52 años, el 65,6% varones, mayoritariamente de raza blanca y con un tercio de los pacientes obesos. El polimorfismo Q80K estaba presente al inicio en el 13,1% de los pacientes del estudio (sólo 1 estaba infectado por el genotipo 1b) y en el 30,7% de los pacientes con genotipo 1a. De acuerdo a la escala METAVIR, el 35,1% de los pacientes tenían F0-F1, el 34,3% tenían F2, el 15,4% F3 y el 15,2% F4.
En el análisis por intención de tratar, SMV en combinación con PEG/RBV fue superior a PBO en combinación con PEG/RBV. El 79.2% de los pacientes del brazo de SMV alcanzaron RVS12 en comparación con el 36,8% en el brazo de PBO (Tabla 4). Los resultados de RVS24 fueron similares (78,3% vs. 31,3%).
Los datos de eficacia de SMV fueron consistentes en todos los subgrupos analizados (carga basal, sexo, polimorfismo IL28B, Metavir).
Al igual que los estudios anteriores, en los pacientes con genotipo 1a y polimorfismo Q80K presente tratados con SMV se observó una menor tasa de RVS12 (46,7%) que en los pacientes sin este polimorfismo (78,5%). En este estudio, en los pacientes con genotipo 1a y polimorfismo Q80K, no se observaron diferencias estadísticamente significativas entre los pacientes tratados con PBO y SMV. La RVS12 en pacientes con VHC genotipo 1b tratados con SMV fue de 85,9% frente al 43% en el grupo PBO.
De acuerdo a los criterios de TGR, 92,7% de los pacientes del brazo de SMV recibieron un total de 24 semanas de tratamiento con PEG/RBV, de los cuales 83% alcanzaron RVS12.
| PROMISE | ||
| Pacentes pretratados (Recidiva) | ||
| SMV | PBO | |
| N=260 | N=133 | |
| RVS12 | N=260 | N=133 |
| % | 206/260 | 49/133 |
| Diferencia | 43,0% (IC 33,8-;52,3) p<0,001 | |
Tabla 4. Resumen de la variable principal (RVS 12 ) del estudio PROMISE en pacientes con recidiva (Análisis ajustado por ITT)
Estudio C206 (ASPIRE)
Estudio de fase 2b multicéntrico, aleatorizado, controlado con placebo, doble ciego, de 7 brazos, para evaluar la eficacia y seguridad de SMV 100 ó 150 mg/día durante 12, 24 ó 48 semanas en combinación con interferón pegilado alfa 2a 180 μ g subcutáneo y RBV 1000-1200 mg/día durante 48 semanas (Figura 1).
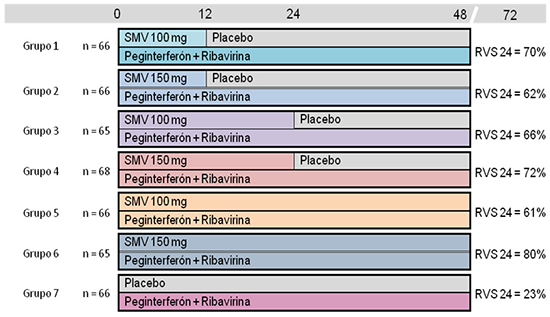
Figura 1. Resultado del tratamiento en pacientes adultos con infección por el genotipo 1 del VHC con fracaso previo a PEG + RBV - Estudio C206 (Análisis ajustado por ITT)
Se incluyeron pacientes con infección activa por VHC con genotipo 1 que habían recaído o fracasado (respondedores parciales o nulos) a un tratamiento previo basado en interferón. Los pacientes debían carecer de antecedentes de descompensación hepática y no estar coinfectados por VHB o VIH.
Se incluyeron 462 pacientes con una edad media de 50 años, 67% varones, mayoritariamente de raza blanca y con un cuarto de los pacientes obesos. Un 41% tenía genotipo 1a y 58% genotipo 1b. El polimorfismo Q80K estaba presente al inicio en el 27% de los pacientes con genotipo 1a. De acuerdo a la escala METAVIR, el 63% de los pacientes tenían F0-F1-F2, el 19% F3 y el 18% F4. Un 40% eran pacientes con recidiva, un 35% respondedores parciales y un 25% respondedores nulos a un tratamiento previo basado en interferón. En total, independientemente de la duración del tratamiento con SMV recibida (12, 24 ó 48 semanas), 199 pacientes recibieron la dosis seleccionada de SMV (150 mg/día), de los cuales 60 recibieron la pauta de SMV finalmente autorizada (150 mg/día durante 12 semanas).
El objetivo principal fue comparar la eficacia y seguridad del tratamiento de SMV + PEG + RBV frente a PEG + RBV. La variable principal del estudio se definió como el porcentaje de pacientes con respuesta viral sostenida en la semana 24 postratamiento (RVS24).
Los resultados del análisis por intención de tratar en los pacientes no respondedores se muestran en la figura 1 para cada uno de los brazos y en la tabla 5 agrupando pacientes que recibieron SMV 150 mg/día durante 12 semanas y SMV 150 mg/día (independientemente de si la duración de tratamiento con SMV fue 12, 24 o 48 semanas).
Las tasas de RVS24 fueron más altas en los pacientes tratados con SMV que en los que recibían placebo en combinación con PEG y RBV, independientemente de la dosis y de la duración del tratamiento. El porcentaje de respuesta en los pacientes en tratamiento con la pauta autorizada de 150 mg durante 12 semanas fue 72,9% y 22,7% en los pacientes con placebo. Los datos de eficacia de SMV fueron consistentes en los subgrupos analizados.
| SMV 150 mg/día 12 semanas % (n/N) | SMV 150 mg/día (12, 24 ó 48 semanas) % (n/N) | PBO % (n/N)) | |
| RVS24 | |||
| Respondedores parciales | 65% (15/17) | 75% (52/69)1 | 9% (2/23) |
| Cirrosis (F4) | - | 82% (9/11) | 0% (0/2) |
| Respondedores nulos | 53% (9/17) | 51% (26/51)2 | 19% (3/16) |
| Cirrosis (F4) | - | 31% (4/13) | 0% (0/2) |
Tabla 5. Resultados del tratamiento en pacientes adultos con infección por el genotipo 1 del VHC con fracaso previo a PEG+ RBV agrupado por dosis - Estudio C206 (Análisis ajustado por ITT) 1 p < 0,001; 2 p = 0,001.
Estudio ATTAIN
Estudio de fase 3 multicéntrico, doble ciego, aleatorizado, para investigar la eficacia de SMV frente a TVR durante 12 semanas en combinación con interferón pegilado y RBV 48 semanas (13).
Se incluyeron pacientes con infección activa por VHC con genotipo 1 que habían recaído o fracasado (respondedores parciales o nulos) a un tratamiento previo basado en interferon. Los pacientes debían carecer de antecedentes de descompensación hepática y no estar coinfectados por VHB o VIH.
Se incluyeron un total de 763 pacientes. Las características basales de la población en ambos brazos fueron similares, con una edad media de 50/52 años (SMV/TVR respectivamente), 64%/58% varones, mayoritariamente de raza blanca. Un 42%/43% tenía genotipo 1a y un 57%/57% genotipo 1b. El polimorfismo Q80K estaba presente al inicio en el 23% de los pacientes con genotipo 1a en el brazo de SMV. De acuerdo a la escala METAVIR, un 29,3%/27,4% de los pacientes tenían F0-F1, un 26,4%/28% tenían F2, un 26%/29% F3 y 18%/15,4% F4. Un 62% eran pacientes respondedores nulos a un tratamiento previo basado en interferón y 38% respondedores parciales.
El objetivo principal fue comparar la eficacia y seguridad del tratamiento de SMV + PEG + RBV frente a TVR + PEG + RBV. La variable principal del estudio se definió como el porcentaje de pacientes con respuesta viral sostenida en la semana 12 postratamiento (RVS 12 ).
Los resultados del análisis por intención de tratar en los pacientes no respondedores se muestran en la tabla 6 para cada uno de los brazos.
| Respondedores Nulos | Respondedores Parciales | |||
| SMV | TVR | SMV | TVR | |
| N=234 | N=238 | N=145 | N=146 | |
| RVS12 | 102/234 | 110/238 | 101/145 | 100/148 |
| % | 43,6% | 46,2% | 69,7% | 68,5% |
| Diferencia | -2,8% (IC95% -11,3-5,8) p<0,001 | 1,5 % (IC95% -9-12) p<0,001 | ||
| RVS12 | 15/61 | 15/48 | 15/27 | 14/27 |
| % cirrosis (F4) | 24,6% | 31,3% | 55,6% | 51,9% |
Tabla 6. Resumen de la variable principal (RVS 12 ) del estudio ATTAIN en pacientes no respondedores (Análisis ajustado por ITT)
Los datos de eficacia de SMV fueron consistentes en todos los subgrupos analizados (carga basal, sexo, polimorfismo IL28B, Metavir).
Genotipo 4 en pacientes sin tratamiento previo y pretratados
Estudio HPC3011 (RESTORE)
Estudio de fase III, abierto, con un solo grupo en pacientes con infección por el genotipo 4 no tratados previamente o con fracaso del tratamiento previo con PEG/RBV (incluidos los pacientes con recidiva previa y con respuesta parcial y nula previas). Los pacientes no tratados previamente (naïve) o con recidiva previa recibieron tratamiento una vez al día con 150 mg de SMV en combinación con PEG y RBV durante 12 semanas, seguido de 12 ó 36 semanas de tratamiento con PEG/RBV de acuerdo con los criterios de TGR. Los pacientes no respondedores (respuesta parcial y nula) recibieron tratamiento una vez al día con 150 mg de SMV en combinación con PEG y RBV durante 12 semanas, seguido de 36 semanas de PEG/RBV.
Los pacientes debían carecer de antecedentes de descompensación hepática y no estar coinfectados por VHB o VIH.
El objetivo principal fue la eficacia del tratamiento con SMV + PEG + RBV definida como porcentaje de pacientes con RVS12.
Se incluyeron 107 pacientes con una edad media de 49 años, 79% varones, mayoritariamente de raza blanca (72%) y con 14% de los pacientes obesos. Ninguno de los pacientes tenía el polimorfismo Q80K presente al inicio. Según la escala METAVIR, el 57% de los pacientes tenían F0-F1-F2, el 14% F3 y el 29% F4. El 42% tenían el genotipo 4a del VHC y el 24% el genotipo 4d del VHC. El 33% (n=35) eran pacientes no tratados previamente, un 21% (n=22) pacientes con recidiva previa, un 9% (n=10) pacientes con respuesta parcial y un 37% (n=40) pacientes con respuesta nula previa.
En el análisis por intención de tratar, la tasa de RVS12 global fue del 65% (70/107); las tasas de RVS12 fueron 83% (29/35) en los pacientes sin tratamiento previo, el 86% en los pacientes con recidiva previa (19/22), el 60% (6/10) en los pacientes con respuesta parcial y el 40% (16/40) en los pacientes respondedores nulos.
Régimen libre de interferón
Estudio HPC2002 (COSMOS)
Estudio abierto, aleatorizado, fase IIa, para investigar la eficacia y seguridad de 12 ó 24 semanas de tratamiento con SMV (150 mg una vez al día) en combinación con SOF (400 mg una vez al día) con o sin RBV en pacientes infectados por el genotipo 1 del VHC en dos cohortes distintas. La cohorte 1 estaba constituida por pacientes respondedores nulos a un tratamiento previo basado en interferón y tenían una fibrosis F0-F2 según la escala METAVIR y la cohorte 2 por pacientes naïve o con respuesta nula previa a un tratamiento previo basado en interferón con fibrosis F3-F4 según la escala METAVIR y enfermedad hepática compensada (Figura 2).

Figura 2. Estudio HPC2002 (COSMOS); análisis ajustado por ITT
En la cohorte 1, se incluyeron 80 pacientes con una edad media de 54 años. El 61% eran varones, mayoritariamente blancos (71%) y aproximadamente un tercio de los pacientes eran obesos. Según la escala METAVIR, 41% tenían fibrosis F0-F1 y 59% tenían F2; el 78% tenía genotipo 1a. El polimorfismo Q80K estaba presente al inicio en el 39% de todos los pacientes y en 50% de los pacientes con genotipo 1a. Todos los pacientes eran respondedores nulos previos a PEG/RBV.
En la cohorte 2, se incluyeron 87 pacientes con una edad media de 57 años. El 67 % eran varones, mayoritariamente blancos y un 44% obesos. Según la escala METAVIR, un 53% tenían F3 y un 47% F4. El 78% tenía genotipo 1a. El polimorfismo Q80K estaba presente al inicio en 31% de todos los pacientes y en el 40% de los pacientes con genotipo 1a. Un 54% de los pacientes eran respondedores nulos y 46% eran pacientes naïve.
Los resultados del análisis por intención de tratar de los pacientes con respuesta nula previa en la cohorte 1 y de los pacientes naïve y con respuesta nula previa en la cohorte 2 para las distintas combinaciones y duraciones de tratamiento se muestran en la figura 2.
Las tasas de RVS12 en los grupos de tratamiento que recibieron 24 semanas de SMV combinado con SOF con y sin RBV fueron similares a las de los pacientes que recibieron 12 semanas. Las tasas de RVS12 en función del geno/subtipo del VHC y del polimorfismo basal Q80K fueron similares en los diferentes subgrupos. Se notificaron un total de 6 pacientes con recidiva viral (6/167, 3,5%): cuatro ocurrieron en pacientes con genotipo 1a y polimorfismo basal Q80K (3 en la cohorte 1 y uno en la cohorte 2) y dos en pacientes con genotipo 1a sin polimorfismo Q80K.
Simeprevir en poblaciones especiales (coinfectados VIH/VHC)
Estudio C212
Estudio de fase III abierto, multicéntrico, con un solo brazo en pacientes con VIH-1 coinfectados con el genotipo 1 del VHC naive o con fracaso al tratamiento previo con PEG/RBV (incluidos pacientes con recidiva previa, respuesta parcial o respuesta nula). Los pacientes naive o con recidiva previa no cirróticos recibieron 12 semanas de tratamiento una vez al día con 150 mg de SMV más PEG alfa-2a y RBV, seguido de 12 ó 36 semanas de tratamiento con peginterferón alfa-2a y ribavirina en función de la TGR. Los pacientes sin respuesta previa (respuesta parcial y respuesta nula) y todos los pacientes cirróticos (grado de fibrosis F4 según la escala METAVIR) recibieron 36 semanas de PEG/RBV después de las 12 semanas iniciales de SMV en combinación con PEG/RBV.
El objetivo principal fue la eficacia del tratamiento con SMV + PEG + RBV definida como porcentaje de pacientes con respuesta viral sostenida en la semana 12 postratamiento (RVS12).
Se incluyeron 106 pacientes con una edad media de 48 años, 85% varones, mayoritariamente de raza blanca y un 12% de pacientes obesos. De acuerdo a la escala METAVIR, el 68% de los pacientes tenían F0-F1-F2, el 19% F3 y el 13% F4. El 82% tenían genotipo 1a y el 17% genotipo 1b. Un 28% de la población total y un 34% de los pacientes con el genotipo 1a tenían el polimorfismo basal Q80K. El 50% (n=53) eran pacientes no tratados previamente frente el VHC, el 14% (n=15) había tenido recidiva previa, el 9% (n=10) eran pacientes con respuesta parcial previa y el 26% (n=28) eran pacientes con respuesta nula previa.
En el análisis por intención de tratar, el 79% de los pacientes no tratados previamente, el 87% (13/15) de los pacientes con recidiva previa, el 70% (7/10) de los pacientes con respuesta parcial y el 57% (16/28) de los pacientes con respuesta nula previa alcanzaron RVS12.
De acuerdo a los criterios de TGR, el 89% de los pacientes no tratados previamente y o con recidiva previa sin cirrosis recibieron un total de 24 semanas de tratamiento con PEG/RBV, de los cuales, el 87% alcanzaron RVS12.
Presencia basal del polimorfismo Q80K
Las tasas de RVS en los pacientes con genotipo 1a y polimorfismo Q80K basal presente fueron sistemáticamente inferiores en todos los estudios (en comparación con los pacientes que no presentaban este polimorfismo), siendo incluso comparables las del grupo placebo en el estudio C205.
Seguridad
Los datos de seguridad de SMV se basan en los 1846 pacientes y 806 voluntarios sanos incluidos en los estudios de fase I, II, III y los estudios en poblaciones especiales, de los cuales un total de 1.153 pacientes recibieron las dosis y duración de tratamiento autorizados.
En los datos agrupados de seguridad de los estudios fase 3, la mayoría de las reacciones adversas notificadas durante las 12 semanas de tratamiento con SMV fueron de intensidad grado 1 ó 2. Se notificaron reacciones adversas de grado 3 ó 4 en el 20% de los pacientes que recibieron SMV con PEG + RBV, frente al 21,9% de los pacientes que recibieron PBO con PEG+RBV. Los efectos adversos graves (EAG) fueron muy raros. Se notificaron reacciones adversas graves en el 0,3% de los pacientes tratados con SMV (2 acontecimientos de fotosensibilidad necesitaron hospitalización) y en ninguno de los que pacientes que recibieron PBO con PEG + RBV. Se notificaron un total de 4 muertes, ninguna de ellas se consideró que tenían relación con SMV.
Durante las primeras 12 semanas de tratamiento, las reacciones adversas notificadas con mayor frecuencia (incidencia ≥5%) fueron náuseas (22%), exantema y prurito (22%), disnea (11,8%), aumento de la bilirrubina en sangre (7,4%) y reacción de fotosensibilidad (5%).
Durante las 12 semanas de tratamiento con SMV, se notificó “elevación de la bilirrubina en sangre” en el 7,4% de los pacientes tratados con simeprevir, en comparación con el 2,8% de los pacientes tratados con PBO con PEG/RBV (todos los grados; datos agrupados de los estudios fase 3). En el 2% y 0,3% de los pacientes tratados con SMV se notificó “elevación de la bilirrubina en sangre” de grado 3 ó 4, respectivamente (datos agrupados de los estudios fase 3). La interrupción de SMV debido a “elevación de la bilirrubina en sangre” fue rara (0,1%; n=1). Durante la administración de SMV con PEG + RBV, las elevaciones de la bilirrubina directa e indirecta no se asociaron generalmente con elevaciones de las transaminasas hepáticas y se normalizaron después del final del tratamiento.
Durante las 12 semanas de tratamiento con SMV, se observó exantema y prurito en el 21,8% y 21,9% de los pacientes tratados con SMV, en comparación con el 16,6% y el 14,6% de los pacientes tratados con PBO. La mayoría de los episodios de exantema y prurito fueron de intensidad leve o moderada (grado 1 ó 2). El 0,5% y el 0,1% de los pacientes tratados con SMV sufrieron exantema o prurito de grado 3, respectivamente. El 0,8% y el 0,1% de los pacientes tratados con SMV, interrumpieron el tratamiento con SMV debido a exantema o prurito, en comparación con el 0,3% y el 0% de los pacientes tratados con PBO/PEG/RBV.
Las abandonos por efectos adversos fueron muy bajos en todos los estudios analizados, 2,6% en todos los brazos que incluyeron SMV durante 12 semanas y 4,5% en los brazos de PEG + RBV. Según los datos agrupados de los estudios de fase 3, se interrumpió la administración de SMV por reacciones adversas en el 0,9% de los pacientes que recibían SMV con PEG + RBV.
No hubo diferencias significativas en las alteraciones en los parámetros de laboratorio observadas en los pacientes tratados con SMV en comparación con los tratados con placebo, peginterferón alfa y ribavirina, a excepción del aumento de bilirrubina en sangre
De acuerdo a los resultados de los estudios, el perfil de seguridad de SMV es similar en los pacientes con infección por el genotipo 4 del VHC y en pacientes co-infectados por el VIH.
No se identificaron nuevos hallazgos sobre la seguridad de SMV en combinación con SOF con o sin RBV más allá de los observados en el tratamiento combinado de SMV con PEG+RBV.
En los pacientes con insuficiencia hepática grave tratados con SMV+PEG+RBV, la exposición de SMV es significativamente mayor. No existen datos en pacientes Child-Pugh B o C. Los aumentos en los niveles de bilirrubina no estaban asociados con ningún hallazgo adverso de seguridad en el hígado.
Se han observado aumentos de los niveles de SMV en pacientes con insuficiencia renal avanzada y no existen datos clínicos en estos pacientes, incluidos los que precisan hemodiálisis.
También se notificaron tasas más altas de anemia en pacientes con fibrosis avanzada.
Interacciones con otros medicamentos
No se recomienda la administración concomitante de SMV con sustancias que moderada o potentemente inducen o inhiben el citocromo P450 3A (CYP3A4) ya que puede originar una exposición a SMV notablemente inferior o superior, respectivamente.
DISCUSIÓN
SMV en combinación con PEG + RBV ha demostrado ser más eficaz que la biterapia en pacientes genotipo 1 naïve y en pacientes tratados previamente con biterapia, con un perfil de efectos adversos no muy diferente al de PEG + RBV. Se administra una vez al día durante 12 semanas, junto con PEG + RBV durante 24 o 48 semanas.
Es importante destacar que los pacientes con enfermedad hepática en estadios avanzados están escasamente representados, así como los pacientes mayores de 65 años y, por tanto, las conclusiones en este grupo de pacientes son menos sólidas.
A continuación se resaltan algunos aspectos importantes a considerar sobre el tratamiento con simeprevir.
Genotipo 1
Los estudios presentados demuestran la eficacia y seguridad de SMV+PEG/RBV durante 12 semanas en pacientes infectados por VHC genotipo 1 naïve y en pacientes que habían recaído tras haber recibido un tratamiento previo basado en interferón.
Un aspecto fundamental que impacta en la eficacia del SMV es la presencia del polimorfismo Q80K, tal y como muestran los análisis de virología realizados en los ensayos clínicos en pacientes no tratados previamente y pre-tratados. Las tasas de RVS fueron más bajas en pacientes con genotipo 1a cuando el polimorfismo está presente e incluso sin diferencias significativas con el grupo placebo, con mayores tasas de recaídas y de fracaso durante los tratamientos. En pacientes infectados con VHC genotipo 1a, la presencia basal del polimorfismo tiene un impacto claro sobre la respuesta virológica a simeprevir, por lo que no se recomienda el uso de SMV en estos casos y deben considerarse tratamientos alternativos.
En el momento actual, las alternativas de tratamiento en pacientes con genotipo 1 incluyen la triple terapia con un inhibidor de proteasa (TVR o BOC) o con SOF y la biterapia SOF con RBV en pacientes con contraindicación a utilizar IFN (14-19). Cuando se diseñaron los estudios con SMV el tratamiento estándar era la biterapia con PEG/RBV por lo que la mayoría de los estudios no se han realizado en comparación con los inhibidores de la proteasa de primera generación ni con sofosbuvir. El ensayo ATTAIN compara directamente SMV con telaprevir en pacientes no respondedores a terapia previa con PEG/RBV.
En pacientes naïve, la triple terapia con TVR o BOC en combinación con PEG+RBV, eleva las tasas de RVS hasta el 75% en cifras globales, si bien es cierto que la coadministración de TVR o BOC con PEG/RBV se asocia con un aumento de la tasa y la gravedad de los acontecimientos adversos, tales como anemia y exantema grave, en comparación con PEG/RBV, especialmente en pacientes con enfermedad hepática más avanzada, lo que no se ha observado en los ensayos clínicos con SMV.
La terapia con SMV supone unas tasas de RVS comparables a las alcanzadas con PEG/RBV + boceprevir o telaprevir (80-85% frente a 67-79%, respectivamente). Cuando se aplican criterios de tratamiento guiado en función de la respuesta, los pacientes que presentan una respuesta virológica rápida extendida (RVRe), es decir, ARN del VHC indetectable en la semana 4 y 12 tienen tasas de RVS elevadas y comparables entre los tres medicamentos (90% vs 96,3% y 92%, respectivamente). Sin embargo, la utilización de SMV aumenta el porcentaje de pacientes que alcanzan una RVRe respecto a TVR/BOC, permitiendo acortar la duración del tratamiento a 24 semanas en aproximadamente un 90% de los pacientes naïve y recaedores.
Un meta-análisis reciente, donde se lleva a cabo una comparación indirecta entre los cuatro antivirales de acción directa autorizados hasta el momento, estima las siguientes tasas globales de RVS12 en pacientes naïve: SMV en pacientes sin polimorfismo Q80K (84%), SOF (83%), BOC (73%) y TVR (74%). Reconociendo las limitaciones de las comparaciones indirectas, éste meta-análisis concluye que, en pacientes con genotipo 1 naïve, SMV y SOF tienen una eficacia comparable entre ellos y superior a la alcanzada con BOC y TLV, permitiendo además un acortamiento de la duración de tratamiento (20).
Los porcentajes de RVS en pacientes con recidiva a un tratamiento previo con PEG/RBV se consideran adecuados y comparables a boceprevir y telaprevir (79,2% frente a 84% y 79%, respectivamente). Los resultados del estudio de fase 3 donde se compara la eficacia de SMV frente a TVR en pacientes con respuesta nula o parcial al tratamiento previo con PEG/RBV indican tasas de respuesta similares en ambos brazos de tratamiento. En el subgrupo de pacientes cirróticos, el porcentaje de RVS fue menor en el brazo de SMV en los pacientes con respuesta nula a un tratamiento previo basado en interferón. Esta diferencia no se observa en el grupo de respondedores parciales. En general, los resultados de este estudio se consideran adecuados para apoyar la pauta de SMV 12 semanas en combinación con PEG/RBV 48 semanas en pacientes que han fracasado a terapia previa con PEG/RBV. En este estudio se observa una incidencia de reacciones adversas y número de abandonos por reacciones adversas menor en el brazo de SMV.
Cuando se comparan las tasas de RVS obtenidas en los brazos con diferentes duraciones de tratamiento de SMV (12, 24 y 48 semanas), los datos de este estudio muestran que prolongar la duración de SMV más allá de las 12 semanas no supone un incremento de la eficacia y que la interrupción de SMV a las 12 semanas, no supone un aumento del número de pacientes con rebotes virológicos o recaídas.
En pacientes infectados con VHC genotipo 1a, en presencia de la mutación Q80K o cuando esta prueba no está disponible, no se recomienda el uso de SMV y deben considerarse tratamientos alternativos.
No existen datos de eficacia/seguridad en pacientes con cirrosis descompensada. Los porcentajes de curación en pacientes con cirrosis parecen inferiores a los pacientes no cirróticos, aunque los datos en esta población son limitados. A pesar de ello, las tasas de respuesta son superiores a los obtenidos con la biterapia y comparables a otros IP.
No se han llevado a cabo estudios en pacientes que hayan fracasado a la terapia con otros antivirales directos
Genotipo 4
La pauta SMV + PEG + RBV ha sido evaluada en un estudio fase III de un solo brazo cuyos resultados son consistentes con los obtenidos en los ensayos clínicos de pacientes infectados con el genotipo 1, apoyando el uso de SMV en el genotipo 4, en las mismas condiciones recomendadas para su uso en el genotipo 1 establecidas en la ficha técnica.
Coinfección VIH/VHC
El uso de SMV+ PEG/RBV en pacientes VIH/VHC presenta resultados similares en eficacia y seguridad a los observados en pacientes monoinfectados en los estudios específicos realizados, disponiéndose de datos en pacientes naïve, con recidiva, respondedores parciales y respondedores nulos. Globalmente, las tasas de RVS obtenidas con esta combinación en esta población de pacientes son superiores a las que se consiguen con PEG/RBV.
Es importante mencionar la existencia de interacciones significativas con algunos antirretrovirales metabolizados por la P450, por tanto, se debe tener en cuenta los medicamentos antirretrovirales indicados en la ficha técnica con los que se puede administrar conjuntamente. En concreto, se debe de evitar el tratamiento simultáneo con inhibidores de la transcriptasa inversa no análogos de nucleósidos (salvo rilpivirina) y con cualquier inhibidor de la proteasa.
Por todo ello, tanto las indicaciones como las estrategias de tratamiento en pacientes coinfectados por VIH/VHC deberían ser idénticas a las de los pacientes mono-infectados. Todo lo referido previamente para cada genotipo en los pacientes mono-infectados aplicaría igual en los pacientes coinfectados VIH/VHC.
Elección del peginterferón alfa
A pesar de que en el estudio QUEST2, los pacientes con terapia triple con PEGinterferon alfa-2a tuvieron una respuesta superior (88%) frente a PEGinterferon alfa-2b (78%), hay que tener en cuenta que se utilizaron con dosis distintas de RVB y que el primero se utilizó en pacientes europeos y el segundo en poblaciones americanas, por lo que no se puede concluir que la eficacia varíe en función del interferón pegilado utilizado.
Reglas de parada
La existencia de una regla de parada a las 4 semanas en pacientes naive o con recidiva supone ventajas frente a los otros inhibidores de la proteasa y SOF.
Régimen libre de interferón
Los resultados del estudio de Fase IIb COSMOS muestran tasas de RVS muy elevadas, que parecen ser independientes de la asociación o no con RBV y de la duración de tratamiento, así como de la presencia de factores de mal pronóstico de respuesta. El tratamiento con SMV + SOF mostró tasas de RVS del 90% permitiendo además acortar la duración del tratamiento hasta las 12 semanas así como una mejora del perfil de seguridad. Debido a que el nivel de evidencia disponible es todavía limitado, el uso de esta combinación con o sin RBV debe limitarse a aquellos pacientes con contraindicaciones para el tratamiento basado en IFN, de acuerdo a las recomendaciones clínicas y a la necesidad de tratamiento. El tiempo más probable de tratamiento en estos casos será de 12 semanas.
Seguridad
En general, SMV presenta un perfil de seguridad mejor que telaprevir y boceprevir y buena tolerancia, con muy pocas interrupciones del tratamiento debidas a acontecimientos adversos y una incidencia baja de acontecimientos adversos graves.
CONCLUSIÓN
Simeprevir es un inhibidor selectivo de la proteasa NS3/4A del VHC, que pertenece a la misma clase farmacoterapéutica, y por consiguiente con un mecanismo de acción e indicación similares, que los otros dos inhibidores de la proteasa (Telaprevir y Boceprevir) ya autorizados la UE para el tratamiento de hepatitis C crónica. A diferencia de éstos, sin embargo, SMV parece tener un mejor perfil de seguridad y ofrece una pauta de tratamiento más cómoda para el paciente.
En consonancia con las consideraciones anteriores, en pacientes infectados con VHC genotipo 1, en base a las tasas de curación observadas y a un perfil de seguridad más favorable que telaprevir y boceprevir, se considera a que simeprevir representa una alternativa terapéutica a los otros dos inhibidores de la proteasa, en pacientes candidatos al tratamiento con triple terapia basada en interferón pegilado + ribavirina. En pacientes naive y con recidiva, la terapia triple con SMV se considera también una alternativa terapéutica a la terapia triple con SOF.
Sin embargo, excepciones a esta recomendación serían aquellos pacientes infectados por VHC genotipo 1a que presenten polimorfismo basal Q80K. En estos pacientes, no se recomienda el uso de simeprevir.
Al igual que sofosbuvir, y a diferencia de otros inhibidores de la proteasa disponibles, simeprevir puede utilizarse en pacientes infectados por el VHC genotipo 4 y en combinaciones libres de interferón. En genotipo 4 debería utilizarse con los mismos criterios de priorización en función de las características individuales de cada paciente, gravedad y grado de fibrosis que para Genotipo 1 con boceprevir y telaprevir.
Simeprevir, en combinación con sofosbuvir +/- RBV durante 12 semanas, se considera una alternativa terapéutica en aquellos pacientes que no toleran o no son candidatos al tratamiento con interferón y existe una necesidad de tratamiento urgente, en los que el tratamiento debe basarse en combinaciones de antivirales directos libres de interferón.
En pacientes en lista de espera de trasplante hepático, trasplantados, pacientes que han fracasado a tratamiento con boceprevir o telaprevir y pacientes con cirrosis avanzada, apenas existen datos de eficacia con SMV. En el caso de pacientes con fracaso a boceprevir o telaprevir, debe considerarse si dicho fracaso ha sido por motivos de seguridad o por recaída tras finalización del tratamiento antes de iniciar el tratamiento con SMV. Los datos con SMV en cirróticos con respuesta nula a biterapia son muy limitados.
REFERENCIAS
1. Guidelines for the screening, care and treatment of persons with hepatitis infection. WHO April 2014. Disponible en: http://apps.who.int/iris/bitstream/10665/111747/1/9789241548755_eng.pdf?ua=1&ua=1
2. EASL Recommendations on Treatment of Hepatitis C 2014. Disponible en: http://files.easl.eu/easl-recommendations-on-treatment-of-hepatitis-C/index.html
3. Criterios y recomendaciones generales para el tratamiento con boceprevir y telaprevir de la hepatitis crónica C en pacientes co- infectados por el VIH y trasplantados hepáticos. Disponible en: http://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-criterios-VHC-co-infectados.pdf
4. Berenguer J, Álvarez-Pellicer J, Martin PM, et al. Sustained virological response to interferon plus ribavirin reduces liver-related complications and mortality in patients coinfected with human immunodeficiency virus and hepatitis C virus. Hepatology. 2009;50(2):407-413.
5. Charlton MR, Gane E, Manns M, et al. Sofosbuvir and Ribavirin for the Treatment of Established Recurrent Hepatitis C Infection After Liver Transplantation: Preliminary Results of a Prospective, Multicenter Study. Hepatology: Special Issue: The 64th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting 2013. 2013;Vol 58(4):1378A.
6. Coilly A, Roche B, Dumortier J, et al. Safety and efficacy of protease inhibitors to treat hepatitis C after liver transplantation: A multicenter experience. J.Hepatol. 2014;60(1):78-86.
7. Crippin JS, McCashland T, Terrault N, Sheiner P, Charlton MR. A pilot study of the tolerability and efficacy of antiviral therapy in hepatitis C virus-infected patients awaiting liver transplantation. Liver Transpl. 2002;8(4):350-355.
8. Curry MP, et al. Pretransplant Sofosbuvir and Ribavirin to Prevent Recurrence of HCV Infection after Liver Transplantation. 64th Annual Meeting of the American Association for the Study of Liver Disease. Nov 1-5, 2013, 2013; Washington, DC.
9. Di Bisceglie A, Kuo A, Rustgi V, Sulkowski M. Virological Outcomes and Adherence to Treatment Algorithms in a Longitudinal Study of Patients with Chronic Hepatitis C Treated with Boceprevir or Telaprevir in the U.S. (HCV-TARGET). AASLD The Liver Meeting. Nov 1-5, 2013, 2013; Washington, DC; 2013.
10. Forns X, Fontana RJ, Moonka D, et al. Initial Evaluation of the Sofosbuvir Compassionate Use Program for Patients with Severe Recurrent HCV Following Liver Transplantation. Special Issue: The 64th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting 2013. Epub Oct 1, 2013, 10-1- 2013a;732A.
11. Ficha técnica de Simeprevir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002777/WC500167867.pdf
12. EPAR de Simeprevir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002777/WC500167870.pdf
13. Reddy KR. et al. A Phase III randomised, double-blind study to evaluate the efficacy, safety and tolerability of simeprevir vs telaprevir in combination with pegylated interferon and ribavirin in chronic hepatitis C virus genotype 1 treatment-experienced patients: the ATTAIN study. 24th Conference of the Asian Pacific Association for the Study of the Liver. Disponible en: http://www.natap.org/2014/APASL/APASL_20.htm">http://www.natap.org/2014/APASL/APASL_20.htm">http://www.natap.org/2014/APASL/APASL_20.htm
14. Ficha Técnica de Sofosbuvir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002798/WC500160597.pdf
15. EPAR de Sofosbuvir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002798/WC500160600.pdf
16. Ficha técnica de Telaprevir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf
17. EPAR de Telaprevir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002313/WC500115532.pdf
18. Ficha técnica de Boceprevir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002332/WC500109786.pdf
19. EPAR de Boceprevir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002332/WC500109789.pdf
20. The Comparative Clinical Effectiveness and Value of Simeprevir and Sofosbuvir in the Treatment of Chronic Hepatitis C Infection. Institute for Clinical and Economic Review. April 2014. Disponible en: http://ctaf.org/sites/default/files/assessments/CTAF_Hep_C_Apr14_final.pdf
GRUPO DE EXPERTOS
(por orden alfabético)
Agencia Española de Medicamentos y Productos Sanitarios
Centro de Información Farmacoterapéutica del Servizo Galego de Saúde
Comité de Evaluación de Nuevos Medicamentos en el Ámbito Hospitalario de Euskadi
Grupo Técnico de Utilidad de Medicamentos Hospitalarios de la Comunidad de Madrid
Programa de Armonización Farmacoterapéutica del Servicio Catalán de la Salud
Los miembros del Consejo asesor y de la Comisión farmacoterapéutica en el ámbito hospitalario pueden consultarse en la página web www.gencat.cat/catsalut
Arantxa Sancho López
Servicio de Farmacología Clínica. Hospital Universitario Puerta de Hierro Majadahonda, Madrid. Miembro del Comité de Medicamentos de Uso Humano (CHMP) de la EMA.
Grupo de Expertos en Hepatitis C:
Rafael Bañares Cañizares
Servicio de Aparato Digestivo (Sección de Hepatología). Hospital General Universitario Gregorio Marañon, Madrid
Juan Berenguer Berenguer
Unidad de Enfermedades Infecciosas/VIH. Hospital General Universitario Gregorio Marañón de Madrid
José Luis Calleja Panero
Servicio de Gastroenterología (Unidad de Hepatología). Hospital Universitario Puerta de Hierro Majadahonda
Rafael Esteban Mur
Servicio de Hepatología. Hospital Vall d'Hebron, Barcelona
Xavier Forns Bernhardt
Servicio de Hepatología. Hospital Clinic de Barcelona, Ciberehd, IDIBAPS
Juan González García
Servicio de Medicina Interna (Unidad de VIH). Hospital Universitario La Paz de Madrid
María Luisa Montes Ramírez
Servicio de Medicina Interna (Unidad VIH). Hospital Universitario La Paz, Madrid
Antonio Olveira Martín
Servicio de Aparato Digestivo (Unidad de Hepatología). Hospital Universitario La Paz, Madrid
Martín Prieto Castillo Unidad de Hepatología. Servicio de Medicina Digestiva. Hospital Universitario y Politécnico La Fe, Valencia
Manuel Romero Gómez
Unidad de Gestión Médico-Quirúrgica de Enfermedades Digestivas y ciberehd. Hospital Universitario de Valme. Universidad de Sevilla
Belén Ruiz Antorán
Servicio de Farmacología Clínica. Hospital Universitario Puerta de Hierro Majadahonda
Maria Jesús Téllez Molina
Servicio de Medicina Interna (Unidad de VIH). Hospital Clínico San Carlos de Madrid
Miguel Angel von Wichmann de Miguel
Unidad de Enfermedades Infecciosas. Hospital Donostia (San Sebastián)
Todos los expertos han realizado una declaración de conflictos de interés.
El Laboratorio Titular, la Sociedad Española de Trasplante Hepático, la Asociación Española para el Estudio del Hígado, la Federación Nacional de Enfermos y Trasplantados Hepáticos y la Federación Española de Hemofilia han tenido oportunidad de enviar comentarios al documento, si bien el texto final es el adoptado por el GCPT.
Fe de erratas
Con fecha 20 de noviembre de 2014, se han corregido las siguientes erratas:
- En las Referencias, la tercera referencia, donde decía
Criterios y recomendaciones generales para el tratamiento con boceprevir y telaprevir de la hepatitis crónica C en pacientes infectados por el VIH, en trasplantados de hígado y en población pediátrica. Disponible en:
http://www.aemps.gob.es/medicamentosUsoHumano/medSituacionesEspeciales/docs/criterios-VHC-off-label.pdf, debe decir:
Criterios y recomendaciones generales para el tratamiento con boceprevir y telaprevir de la hepatitis crónica C en pacientes co- infectados por el VIH y trasplantados hepáticos. Disponible en:
http://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-criterios-VHC-co-infectados.pdf

