Informe de Posicionamiento Terapéutico de ledipasvir/sofosbuvir (Harvoni®)
PT-LEDIPASVIR/SOFOSBUVIR/V1/20032015
Informe de Posicionamiento Terapéutico de ledipasvir/sofosbuvir (Harvoni®)
Fecha de publicación: 20 de marzo de 2015
La infección por el virus de la hepatitis C (VHC) es un problema de salud de primera magnitud en Europa y especialmente en los países mediterráneos, donde las tasas de prevalencia oscilan entre el 1-3% (1). Es la primera causa de enfermedad hepática terminal y una de las principales indicaciones de trasplante hepático. La recurrencia de la infección en el órgano trasplantado y un curso más agresivo y acelerado hacen que los resultados a medio plazo del trasplante hepático sean peores que los observados en cirrosis de otras etiologías.
El VHC se divide en 7 genotipos con numerosos subtipos, siendo el genotipo 1 el más frecuente en Europa con una prevalencia aproximada del 70%, seguido del genotipo 3. En España, el genotipo 4 es el tercero en frecuencia siendo el genotipo 2 el más infrecuente. El genotipo del VHC, si bien no condiciona una evolución clínica diferente de la hepatitis C crónica (HCC), sí tiene un gran impacto en la respuesta al tratamiento.
En el momento actual, los tratamientos aprobados y comercializados para la HCC son interferón alfa pegilado (PEG) (2), ribavirina (RBV), tres inhibidores de la proteasa NS3/4A, boceprevir (BOC) (3,4) y telaprevir (TVR) (5,6), activos sólo frente al genotipo 1 y simeprevir (SMV) activo frente a los genotipos 1 y 4, y sofosbuvir (SOF) y daclatasvir (DCV) inhibidores nucleósidos de la polimerasa NS5B y NS5A respectivamente, con actividad frente a todos los genotipos del VHC (7,8,9,10). De forma general, los pacientes infectados con el VHC con una fibrosis significativa (F2- F4) naïve o que no han respondido a un tratamiento previo deben ser tratados, salvo excepciones, con una de las pautas de antivirales orales sin interferón disponibles en el mercado. En los pacientes con fibrosis F0-F1, el tratamiento se puede diferir y considerar su indicación individualmente. Deberán monitorizarse de manera estrecha y tratarse si se producen cambios relevantes en su evolución o en la progresión de su enfermedad (11). Otros pacientes prioritarios para el tratamiento con antivirales orales incluyen: pacientes en lista de espera de trasplante hepático; Pacientes trasplantados hepáticos con recidiva de la infección en el injerto hepático, independientemente de la existencia o no de complicaciones y del estadio de fibrosis; pacientes que no han respondido a inhibidores de la proteasa de primera generación; trasplantados no hepáticos con una hepatitis C, independiente del estadio de fibrosis hepática y Pacientes con hepatitis C con manifestaciones extrahepáticas clínicamente relevantes del VHC, independiente del estadio de fibrosis hepática.
Todos los tratamientos basados en interferón se asocian a un gran número de efectos adversos y una menor efectividad, especialmente en los pacientes con hepatopatías más avanzadas, lo que limita su aplicabilidad en algunos grupos de pacientes (2, 12- 18). Estos efectos adversos incluyen riesgo de descompensación hepática, sepsis e importante mielosupresión. Los efectos adversos psiquiátricos de tipo ansioso-depresivo y el insomnio también son muy frecuentes (2)
Por lo tanto, la gran cantidad de efectos secundarios y la eficacia limitada en algunas poblaciones de las terapias basadas en más avanzada y en los que han fracasado a tratamientos previos, hacen muy necesarios otros fármacos que sean mejor tolerados y más efectivos (12-17).
En el momento actual existe un enorme desarrollo de la investigación de nuevos fármacos frente al VHC que actúan de manera directa y pertenecen a 4 clases diferentes: los inhibidores de proteasa NS3/4A, los inhibidores de NS5A y los inhibidores de la polimerasa NS5B que pueden ser nucleósidos o no nucleósidos. Todas estas nuevas moléculas se están estudiando en diversas combinaciones (incluyendo o no PEG y/o RBV) entre agentes de las diferentes clases, observándose aumentos muy importantes en la eficacia (12-17).
Una de estas moléculas desarrollada es ledipasvir (LDV), el cual se ha combinado a dosis fijas junto con SOF. El 17 de noviembre de 2014, la EMA aprobó el producto de combinación fija de los medicamentos LDV 90 mg / 400 mg SOF llamado Harvoni®(18,19).
LEDIPASVIR /SOFOSBUVIR (HARVONI®)
Harvoni® ha sido autorizado para el tratamiento de la HCC en adultos. Se presenta en comprimidos recubiertos con película y contiene 90 mg de LDV en combinación con 400 mg de SOF. La dosis recomendada es de 1 comprimido una vez al día por vía oral con o sin alimentos.
Tabla 1: Duración recomendada de tratamiento con Ledipasvir/Sofosbuvir y el uso recomendado de añadir ribavirina en determinados subgrupos.
| Población pacientes* | Tratamiento | Duración |
| Pacientes con HCC de genotipo 1 o 4 | ||
| Pacientes sin cirrosis | Harvoni | 12 semanas -Se puede contemplar la administración durante 8 semanas en los pacientes infectados por el genotipo 1 sin tratamiento previo -Se debe contemplar la administración durante 24 semanas en los pacientes previamente tratados con opciones inciertas de retratamiento |
| Pacientes con cirrosis compensada | Harvoni | 24 semanas -Se puede contemplar la administración durante 12 semanas en los pacientes considerados de bajo riesgo de progresión de la enfermedad clínica y que tienen opciones de retratamiento |
| Pacientes con cirrosis descompensada, pre o post-trasplante hepático. | Harvoni + ribavirina | 24 semanas |
| Pacientes con HCC de genotipo3 | ||
| Pacientes con cirrosis y/o fracaso del tratamiento previo | Harvoni + ribavirina | 24 semanas |
* Incluye pacientes co-infectados con el Virus de la Inmunodeficiencia Humana (VIH).
Farmacología
LDV es un inhibidor específico de la proteína NS5A, esencial tanto para la replicación del ARN, como para el ensamblaje de los viriones del VHC, utilizado en combinación con otros fármacos activos frente al VHC.
SOF es un inhibidor nucleótido de la polimerasa NS5B utilizado en combinación con otros fármacos activos frente al VHC en función del genotipo.
La combinación a dosis fijas de LDV/SOF ha sido autorizada en ficha técnica para el tratamiento de los genotipos 1,3 y 4.
Se desarrolló un programa de estudios clínicos fase I para caracterizar la farmacocinética de SOF, LDV y LDV/SOF. Adicionalmente se dispone de los datos de concentración en plasma de 391 sujetos sanos y 2147 sujetos infectados por el VHC, que recibieron LDV/SOF, SOF + LDV o LDV como agente único.
Se dispone de un total de 22 estudios del desarrollo farmacológico clínico de SOF, 13 estudios en fase I, 5 estudios en fase II y 4 estudios en fase III, los cuales ya han sido presentados en el IPT de SOF.
Se han realizado 19 estudios con LDV como agente único.
Se dispone de 11 estudios del desarrollo farmacológico clínico realizados con la combinación de LDV/SOF (5 de fase I, 3 de fase II y 3 de fase III). (20-22).
Eficacia
Se dispone de 3 estudios de fase III del desarrollo farmacológico clínico de Harvoni® con datos disponibles en un total de 1.950 pacientes con hepatitis C crónica genotipo 1.
Los tres estudios de fase III incluyen un estudio realizado en pacientes naïve no cirróticos (ION 3); un estudio en pacientes naïve cirróticos y no cirróticos (ION 1); y un estudio en pacientes cirróticos y no cirróticos en los que ha fracasado el tratamiento previo con un régimen a base de interferón, incluyendo regímenes que contienen un inhibidor de la proteasa del VHC (ION 2). Los pacientes en estos estudios tenían enfermedad hepática compensada. En todos se evaluó la eficacia de LDV/SOF con o sin ribavirina.
La Respuesta Viral Sostenida (RVS) fue la variable principal para determinar la tasa de curación del VHC que se definió como ARN del VHC inferior al Límite Inferior de Cuantificación (Lower Limit of Cuantification, LLOQ) <25 UI/ml a las 12 semanas después de la interrupción del tratamiento.
ADULTOS NAÏVE SIN CIRROSIS. ION 3. Estudio 0108. Genotipo 1 (23)
Estudio multicéntrico, abierto, fase III que aleatorizó a 647 pacientes no tratados previamente infectados con el genotipo 1 del VHC sin cirrosis a recibir LDV/SOF durante 8 semanas, LDV/SOF + ribavirina durante 8 semanas, o LDV/SOF durante 12 semanas. Tiene como objetivo principal comparar la eficacia de LDV/SOF 8 semanas con o sin RBV y LDV/SOF 12 semanas frente a un control histórico. La variable principal de eficacia fue la RVS a las 12 semanas después de finalizar el tratamiento. La aleatorización se estratificó en función del genotipo del VHC (1a frente a 1b).
Entre los análisis secundarios se plantea el realizar una comparación de no inferioridad de las pauta de LDV/SOF 8 semanas con o sin RBV respecto a LDV/SOF 12 semanas. Se utiliza un margen de no inferioridad para la diferencia de tasas de respuesta del 12%.
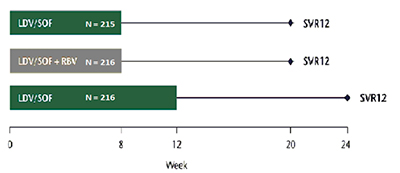
Figura 1: Diseño ION 3
Los 647 pacientes tenían una mediana de edad de 52 años (rango: 20 a 75); el 78% eran de raza blanca y el 19% de raza negra/afroamericanos. El 80% de los sujetos presentaba infección por el genotipo 1a. El 13% de todos los pacientes tenían fibrosis hepática F3 y ningún paciente tenía fibrosis F4. La mayoría de los pacientes (73%) tenían genotipos IL-28B no-CC.
La RVS12 fue del 94% (202/215) con 8 semanas de LDV/SOF, el 93% (201/216) con 8 semanas de LDV/SOF más RBV y el 96% (208/216), con 12 semanas de LDV/SOF. Los tres fueron superiores al control histórico calculado (RVS 12 semanas del 60%).
El brazo de tratamiento de 8 semanas de LDV/SOF sin RBV mostró ser no inferior a los otros dos grupos de tratamiento. En comparación con LDV/SOF 12 semanas la diferencia en RVS12 fue de -1.4% (IC97,5%: -6, a 3,6%) y de 0.9% (IC95%-3,9% a 5,7%) respecto a LDV/SOF + RBV. También se mostró la no inferioridad entre el tratamiento de SOF+LDV + RBV 8 semanas y LDV/SOF 12 semanas (diferencia de -2,3%; IC97,5%: -7,2% a 3,6%).
Tabla 2: Tasas de respuesta. ION-3.
| 8 semanas (n = 215) | 8 semanas (n = 216) | 12 semanas (n = 216) | |
| RVS12 | (IC95%: 90 ─ 97) | (IC95%, 89─96) | (IC95%, 92─98) |
| | |||
| Fallo virológico del tratamiento | | | |
| Recaídas | | | |
| Otros* | | | |
| | |||
| Genotipo 1a | | | |
| Genotipo 1b | | | |
* Otros incluye pacientes que no alcanzaron RVS y no cumplieron criterios de fallo virológico (por ejemplo, pérdidas de seguimiento).
La tasa de recaída es superior cuando la duración del tratamiento se reduce a 8 semanas, independientemente de si se añade RBV al régimen. Estas recaídas se dieron principalmente en varones, con el genotipo IL28 no-CC y / o alta carga viral basal (> 6 millones UI/ml). En los pacientes con > 6 millones UI/ml de carga viral basal, el porcentaje de recaídas se reduce con el tratamiento de 12 semanas (con 8 semanas 8-10% recaídas y con 12 semanas 1%). En pacientes con carga viral baja (<6 millones UI/ml) la tasa de recaídas es del 2% en los 3 brazos de tratamiento.
ADULTOS NAIVE CON O SIN CIRROSIS. ION 1. Estudio 0102. Genotipo 1 (24)
Estudio multicéntrico, fase III, abierto realizado en pacientes no tratados previamente con HCC genotipo 1 (n=865). Los pacientes fueron asignados aleatoriamente en una proporción 1: 1: 1: 1 para recibir LDV/SOF en un comprimido combinado de dosis fija una vez al día durante 12 semanas, LDV/SOF más RBV durante 12 semanas, LDV/SOF durante 24 semanas, o LDV/SOF más RBV durante 24 semanas. La variable principal de eficacia fue la respuesta viral sostenida a las 12 semanas después del final de la terapia. La aleatorización se estratificó en función de la presencia o ausencia de cirrosis y el genotipo del VHC (1a frente a 1b).
Todos los pacientes del ensayo están en tratamiento con LDV/SOF, la eficacia de los grupos se compara frente a un control histórico del 60%.
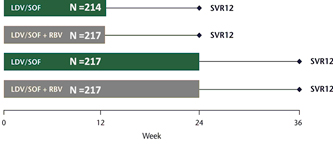
Figura 2: Diseño ION 1.
Los 865 pacientes tenían una mediana de edad de 52 años (rango: 18 a 80); el 85% era de raza blanca y el 12% de raza negra/afroamericanos. El 67% de los sujetos presentaba infección por el genotipo 1a.. El porcentaje de pacientes incluidos con cirrosis hepática fue 15.7% (136/865). La mayoría de los pacientes (70%) tenían genotipos IL-28B no-CC.
Las tasas de RVS12 fueron del 99% en el grupo que recibió 12 semanas de LDV/SOF; 97% en el grupo que recibió 12 semanas de LDV/SOF más RBV; 98% en el grupo que recibió 24 semanas de LDV/SOF y 99% en el grupo que recibió 24 semanas de LDV/SOF más RBV.
El porcentaje de recaídas fue inferior al 1% en ambos brazos (sin o con RBV). El número de pacientes con cirrosis tratados durante 12 semanas (34 con SOF / LDV, 33 con SOF / LDV + RBV) es bajo como para obtener conclusiones definitivas sobre la eficacia relativa de 12 y 24 semanas de tratamiento para este subgrupo; la tasa de RVS12 en este subgrupo de pacientes fue similar con ambas duraciones de tratamiento.
Tabla 3: Tasas de respuesta. ION-1.
| LDV/SOF 12 sem. (n = 214) | (n = 217) | 24 sem. (n = 217) | V 24 sem. (n = 217) | |
| RVS12 | (IC95%: 96─100) | (IC95%, 94─99) | (IC95%, 95─99) | (IC95%, 97─100) |
| | ||||
| Fallo virológico del tratamiento. | | | | |
| Recaída | | | | |
| Otros* | | | | |
| | ||||
| | ||||
| Genotipo 1a | | | | |
| Genotipo 1b | | | | |
| | ||||
| No | | | | |
| Sí | | | | |
* Otros incluye pacientes que no alcanzaron RVS12 y no cumplieron criterios de fallo virológico (por ejemplo, pérdidas de seguimiento).
PACIENTES TRATADOS PREVIAMENTE CON O SIN CIRROSIS. ION 2. Estudio 0109. Genotipo 1 (25)
Estudio multicéntrico, fase III aleatorizado, abierto realizado en pacientes infectados con el genotipo 1 del VHC que no habían tenido una RVS después del tratamiento con peginterferón y RBV, con o sin un inhibidor de la proteasa (n= 440). No se incluyen pacientes adversos. Se permitía la inclusión de pacientes con cirrosis compensada. Los pacientes fueron asignados al azar para recibir LDV/SOF en combinación en un mismo comprimido con dosis fija una vez al día durante 12 semanas, LDV/SOF más RBV durante 12 semanas, LDV/SOF durante 24 semanas, o LDV/SOF más RBV durante 24 semanas. La aleatorización fue estratificada según la existencia o ausencia de cirrosis, genotipo viral (1a o 1b) y respuesta a la anterior terapia. La variable principal de eficacia fue la RVS a las 12 semanas después del fin de la terapia.
Todos los pacientes del ensayo están en tratamiento con LDV/SOF, la eficacia de los grupos se compara frente a un control histórico.
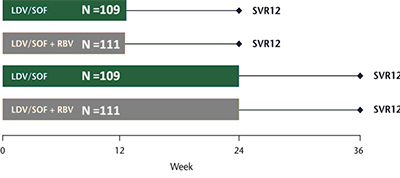
Figura 3: Diseño ION 2
Los 440 pacientes tenían una mediana de edad de 56 años (rango: 24 a 75); el 81% era de raza blanca y el 18% de raza negra/afroamericanos. El 79% de los sujetos presentaba infección por el genotipo 1a. La conversión de la puntuación FibroTest a la puntuación METAVIR correspondiente sugiere que el 58% de todos los pacientes tenían fibrosis hepática ≥F3. El 20 % de los pacientes incluidos tenía cirrosis hepática (88/440). La mayoría de los pacientes (87%) tenían genotipos IL-28B no-CC. El 48% de los sujetos había recibido tratamiento previo basado en PEG/IFN+RBV mientras que el otro 52% había recibido tratamiento con PEG/IFN + RBV y un antiviral inhibidor de la proteasa del VHC.
Las tasas de RVS fueron altas en todos los grupos de tratamiento: 94% en el grupo que recibió 12 semanas de LDV/SOF; 96% en el grupo que recibió 12 semanas de LDV/SOF y RBV; 99% en el grupo que recibió 24 semanas de LDV/SOF; y 99% en el grupo que recibió 24 semanas de LDV/SOF y RBV.
Las recaídas, se observaron únicamente en los brazos de 12 semanas y la mayoría se produjeron dentro de la semana 4 después de suspender la terapia en 10/11 casos.
Se realizó un análisis univariante de la posible relación causal entre las recaídas y distintos factores. De entre ellos, solo se estableció relación significativa como predictores de recaída en la presencia de cirrosis y en tener un recuento de plaquetas basal ≤125,000 / ml.
ANALISIS COMPLEMENTARIOS
Pacientes naïve y tratados previamente con cirrosis compensada
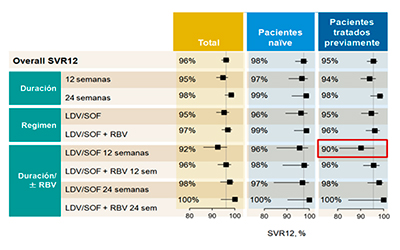
Se han presentado en el congreso americano de noviembre de 2014, los resultados de un análisis integrado de los resultados agregados de los diferentes estudios que incluían un total de 513 pacientes con cirrosis compensada tratados con LDV/SOF con o sin RBV durante 12 ó 24 semanas (Estudio LONESTAR, ELECTRON, ELECTRON-2, 337-0113, ION-1, ION-2, SIRIUS). A pesar de tratarse de un grupo muy heterogéneo de pacientes y que el número de pacientes en los análisis de subgrupos es pequeño en algunos casos (sólo 58 pacientes recibieron LDV/SOF + RBV 24 semanas), este análisis indica que, de forma general, la pauta corta de 12 semanas sin RBV es igual de eficaz que las otras. Sólo se observan resultados de RVS algo inferiores en el subgrupo de pacientes tratados previamente, donde la adición de RBV incrementa la tasa de RVS12 de 90% a 96%) (26).
Tabla 4: Tasas de respuesta ION-2
| 12 sem. (n = 109) | +RBV 12 sem. (n = 111) | 24 sem. (n = 109) | +RBV 24 sem. (n = 111) | |
| RVS | (IC95%; 87─97) | (IC95%, 91─99) | (IC95%, 95─100) | (IC95%, 95─100) |
| | ||||
| Fallo virológico del tratamiento | | | | |
| Recaída | | | | |
| Otros* | | | | |
| | ||||
| | ||||
| Genotipo 1a | | | | |
| Genotipo 1b | | | | |
| | ||||
| No | | | | |
| Sí** | | | | |
| | ||||
| PEG-IFN +RBV | | | | |
| Inhibidor de la proteasa + PEG-IFN +RBV | | | | |
* Otros incluye pacientes que no alcanzaron RVS y no cumplieron criterios de fallo virológico (por ejemplo, pérdidas de seguimiento). ** Metavir score = 4 o Ishak score ≥ 5 por biopsia hepática, o FibroTest score de > 0.75 y (APRI) de> 2.
Tabla 5: Resultados RVS12 integrados pacientes con cirrosis (Estudios LONESTAR, ELECTRON, ELECTRON-2, 337-0113, ION-1, ION-2, SIRIUS)
PACIENTES CON CIRROSIS DESCOMPENSADA y/o POSTRASPLANTE
Estudio ELECTRON-2
El estudio ELECTRON-2 (27,28) es un estudio de fase II, parcialmente aleatorizado, abierto y actualmente en desarrollo.
Este estudio pretende evaluar la eficacia y seguridad de 12 semanas de LDV/SOFcon o sin RBV en pacientes con factores pronósticos de mala respuesta al tratamiento, ya sea con genotipo 3, genotipo 1 y cirrosis descompensada o genotipo 1 y fracaso en la terapia previa con SOF.
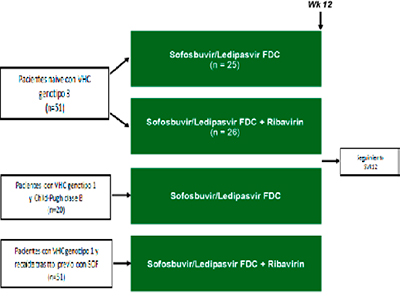
Figura 4: Diseño ELECTRON-2
En el brazo de tratamiento que incluía pacientes con genotipo 1 y Child-Pugh B que recibieron tratamiento con LDV/SOF, se observó una tasa de respuesta virológica sostenida del 65% (13/20). Todos los fracasos del tratamiento se debieron a recaídas (35%, n=7/20).
Estudio SOLAR-1 (29,30)
El estudio SOLAR-1 es un estudio multicéntrico, abierto, actualmente en desarrollo que evalúa la eficacia de la combinación LDV/SOF más RBV en pacientes infectados por VHC genotipos 1 ó 4 con cirrosis descompensada y/o trasplantados hepáticos.
Tabla 6: Diseño SOLAR-1
| Cohorte | Grupo | Daño hepático | Duración de LDV/SOF + RBV | Pacientes tratados |
| Cohorte A - pacientes pre- trasplante con cirrosis descompensada | 1 | CPT B | 12 semanas | 30 |
| 24 semanas | 27 | |||
| 2 | CPT C | 12 semanas | 22 | |
| 24 semanas | 20 | |||
| Total cohorte A | 99 | |||
| Cohorte B - pacientes post- trasplante | 3 | Fibrosis F0-F3 | 12 semanas | 55 |
| 24 semanas | 56 | |||
| 4 | CPT A | 12 semanas | 26 | |
| 24 semanas | 25 | |||
| 5 | CPT B | 12 semanas | 26 | |
| 24 semanas | 18 | |||
| 6 | CPT C | 12 semanas | 5 | |
| 24 semanas | 3 | |||
| 7 | Enfermedad agresiva recurrente | 12 semanas | 4 | |
| 24 semanas | 0 | |||
| Total cohorte B | 218 | |||
El estudio divide a los pacientes en 2 cohortes; A (cirrosis descompensada) y B (post-trasplante), compuestas por 2 y por 5 grupos respectivamente y distribuyendo a los pacientes según el esquema de la tabla 6.
Los datos preliminares del estudio incluyen los resultados de RVS provisionales de un total de 313 pacientes con genotipo 1 y 4. La RVS12 de la cohorte A (pacientes con cirrosis descompensada) es 87% (45/52) LDV/SOF + RBV durante 12 semanas y 89% (42/47) durante 24 semanas. Los resultados por grupos son para el grupo 1 CPT B 87% (26/30) para 12 semanas y 89% (24/27) para 24 semanas. Para el grupo 2 CPT C, 86% (19/22) para 12 semanas y 90% (18/20) para 24 semanas.
En los pacientes postrasplantados de hígado (cohorte B), las tasas de RVS12 con LDV/SOF + RBV fueron: Grupo 3 (Fibrosis F0-F3: 96% (53/55) y 98% (55/56) para 12 y 24 semanas respectivamente. Grupo 4 CPT A, 96% con 12 y 24 semanas (25/26) y (24/25). Grupo 5 CPT B, 85% (22/26) y 83% (15/18) para 12 y 24 semanas respectivamente. Grupo 6 CPT C, 60% (3/5) y 67% (2/3) para 12 y 24 semanas respectivamente.
Tabla 7: Resultados preliminares de RVS12 del estudio SOLAR-1
| Cohorte | Grupo | Daño hepático | Duración de LDV/SOF + RBV | % RVS12 |
| Cohorte A - pacientes pre- trasplante con cirrosis descompensada | 1 | CPT B | 12 semanas | 87% (26/30) |
| 24 semanas | 89% (24/27) | |||
| 2 | CPT C | 12 semanas | 86% (19/22) | |
| 24 semanas | 90% (18/20) | |||
| Total cohorte A | 12 semanas | 87% (45/52) | ||
| 24 semanas | 89% (42/47) | |||
| Cohorte B - pacientes post- trasplante | 3 | Fibrosis F0-F3 | 12 semanas | 96% (53/55) |
| 24 semanas | 98% (55/56) | |||
| 4 | CPT A | 12 semanas | 96% (25/26) | |
| 24 semanas | 96% (24/25) | |||
| 5 | CPT B | 12 semanas | 85% (22/26) | |
| 24 semanas | 83% (15/18) | |||
| 6 | CPT C | 12 semanas | 60% (3/5) | |
| 24 semanas | 67% (2/3) | |||
| 7 | Enfermedad agresiva recurrente | 12 semanas | | |
| 24 semanas | | |||
PACIENTES CON CIRROSIS COMPENSADA QUE HAN FRACASADO A BITERAPIA O TRITERAPIA (Estudio SIRIUS)
Se han presentado en el congreso americano de noviembre de 2014, los resultados de un estudio de fase II, aleatorizado, doble ciego realizado en Francia en el que participaron 155 pacientes con cirrosis compensada, pretratados que no habían conseguido una RVS con PEG +RBV o PEG +RBV+ IP (31). Los pacientes se aleatorizaron para recibir LDV/SOF + RBV durante 12 semanas o LDV/SOF durante 24 semanas. La RVS12 fue del 96% (74/77) en el brazo de LDV/SOF + RBV durante 12 semanas frente al 97% (75/77) con LDV/SOF durante 24 semanas.
PACIENTES CON INFECCION POR VHC GENOTIPO 3
Los datos en pacientes con genotipo 3 provienen del estudio ELECTRON-2 citado anteriormente (27,28).
De los 51 pacientes naïve (figura 4) incluidos, el 100 % (26/26) alcanzaron la RVS12 en el brazo de tratamiento con LDV/SOF + RVB 12 semanas mientras que en el brazo de tratamiento con LDV/SOF 12 semanas sólo lo hicieron el 64 % de ellos (16/25).
Posteriormente fueron proporcionados datos de RVS12 de 50 pacientes más, en este caso pretratados. Todos ellos fueron tratados con LDV/SOF + RBV durante 12 semanas, y se dividieron en cirróticos (n=22) y no cirróticos (n=28). La RVS12 global en los pacientes pretratados fue del 82% (41/50), 89% en no cirróticos (25/28) y del 73% en cirróticos (16/22). Todos estos pacientes recibieron todos LDV/SOF+RBV.
PACIENTES CON INFECCIÓN POR VHC GENOTIPO 4
Dos pacientes con infección por el VHC genotipo 4 fueron incluidos en el estudio ION 1. Un paciente recibió LDV/SOF durante 12 semanas; otro paciente recibió LDV/SOF + RBV durante 24 semanas. Ambos lograron RVS12. En el estudio SYNERGY (CO-US-337-047) de fase II, 21 pacientes naïve y pretratados, tanto cirróticos como no cirróticos, infectados por el VHC genotipo 4 han sido tratados con LDV/SOF durante 12 semanas, de los que han llegado a semana 12 de seguimiento postratamiento 20. 19/20 (95%) de los pacientes han logrado RVS12. LDV y SOF han demostrado actividad antiviral in vitro contra el VHC genotipos 4.
PACIENTES COINFECTADOS POR VHC y VIH
El estudio ION-4 (32) es un estudio de fase III abierto, multicéntrico, que evalúa la eficacia, seguridad y tolerabilidad del tratamiento de LDV/SOF durante 12 semanas en 335 pacientes con genotipo 1a (75%), 1b (23%) o 4 (2%) de VHC y co-infección con VIH-1. El estudio incluyó a pacientes con VHC no tratados con anterioridad (45%) y pacientes tratados previamente (55%). Incluyó además un 20% de pacientes con cirrosis compensada (20%). Respeto al VIH se incluyeron pacientes que estaban recibiendo el siguiente TARGA: tenofovir, entrivitabina, efavirenz; raltegravir o rilpivirina.
La tasa general de RVS12 fue del 96% (321/335). La tasa de RVS12 fue del 95% en pacientes naive y del 97% en pacientes que habían recibido anteriormente un tratamiento frente al VHC. La tasa de RVS12 fue del 96% en pacientes sin cirrosis frente a un 94% en pacientes con cirrosis. De los 14 pacientes que no alcanzaron RVS12, 2 pacientes experimentaron fracaso virológico durante el tratamiento, 10 experimentaron recaída tras la finalización del tratamiento, 1 paciente se perdió durante el seguimiento y 1 paciente falleció por causas no asociadas al tratamiento.
El estudio ERADICATE (33) es un estudio fase II, abierto que evalúa la eficacia de la combinación LDV/SOF en 50 pacientes coinfectados por VHC genotipo 1 y VIH.
Respecto a la infección por el VHC, ningún paciente había recibido tratamiento previo y el grado de fibrosis se encontraba entre 0 y 3. Respecto al VIH se incluyeron pacientes que habían comenzado tratamiento con TARGA (74%) o naive para la misma (26%).
Los cincuenta pacientes tenían una mediana de edad de 58 años, mayoritariamente de raza blanca y con VHC genotipo 1a.
Todos los pacientes recibieron LDV/SOF durante 12 semanas y fueron seguidos posteriormente durante 48 semanas.
Tanto la RVS4 como la RVS12 fue del 100 % (13/13) en los pacientes naïve al tratamiento antirretroviral. En los pacientes que si recibían tratamiento para el VIH, la RVS12 fue del 98 % (36/37). No hubo toxicidad renal ni abandonos por RAM y además no hubo ni cambios significativos en los niveles de CD4.
Seguridad
La tolerabilidad de SOF es buena y no se han atribuido efectos secundarios específicos. La seguridad clínica de LDV ha sido estudiada fundamentalmente en combinación con SOF. Hasta la fecha, no se han notificado efectos secundarios específicos para los inhibidores de NS5A
El perfil de seguridad global de LDV/SOF se basa en datos de 1952 pacientes con infección crónica por el VHC que recibieron LDV/SOF 400mg/90mg una vez al día con (n = 1080) o sin RBV (n= 834) en un total de 3 ensayos clínicos de fase III.
La frecuencia de acontecimientos adversos de grado> 2 fue baja. El número de pacientes que discontinuaron el tratamiento debido a eventos adversos fue muy baja (<1%) siendo los efectos secundarios más comunes el dolor de cabeza leve y la fatiga.
La frecuencia de acontecimientos adversos fue mayor en los pacientes tratados con el régimen triple incluyendo RBV, como se esperaba y son aquellos previamente descritos en el uso de RBV.
La adición de RBV aumentó la frecuencia de acontecimientos adversos de grado 1-2 (el patrón típico para este agente); la frecuencia de acontecimientos adversos de grado 3-4 fue baja al igual que al utilizar SOF / LDV solamente, y no hubo un aumento en interrupciones del tratamiento cuando se añadió al régimen de RBV. Por lo tanto, la adición de RBV para minimizar el riesgo de recaída en ciertos pacientes no está asociada con ningún problema importante de seguridad.
RESISTENCIAS
Respecto a las opciones de tratamiento posterior, las resistencias del NS5A es posible que sean permanentes o de larga duración si presentan resistencia cruzada para todos los miembros de la familia. SOF presenta una aparición de resistencias excepcional y hay datos de que puede ser reutilizado.
DISCUSIÓN
En el momento actual, los tratamientos autorizados para el VHC genotipo 1 incluyen PEG+RBV, co-administrado con un antiviral de acción directa, TVR, BOC, SMV y muy recientemente, SOF y DCV. Estos tratamientos han demostrado ser más eficaces que la combinación de PEG/RBV sola. Con la autorización en 2011 de la terapia triple que añade TVR o BOC a la combinación de PEG+RBV, se elevaron las tasas de respuesta hasta el 75% en cifras globales, si bien es cierto que la coadministración de TVR o BOC con PEG/RBV se asocia con un aumento de la tasa y la gravedad de los acontecimientos adversos, tales como anemia y exantema grave, en comparación con PEG/RBV sola, especialmente en pacientes con enfermedad hepática más avanzada. A diferencia de éstos, SMV, DCV y SOF son activos frente a más genotipos, tienen un mejor perfil de seguridad y permiten acortar la duración del tratamiento
De forma general, es importante mencionar que no existen estudios de comparación directa entre LDV/SOF± RBV con PEG + RBV o PEG + RBV+ IP, ni frente a otras alternativas sin interferón, SOF+DCV o SOF + SMV, lo que implica ciertas limitaciones (principalmente ausencia de grupo control, lo que limita el control de los sesgos de selección, observación y análisis). Esto dificulta la posibilidad de llevar a cabo una comparación indirecta ajustada de los diferentes antivirales directos en regímenes libres de interferón. Sin embargo, dado que los diferentes estudios emplearon la misma variable principal de estudio y las poblaciones son comparables, aunque no sea posible establecer una comparación estadística, si nos permiten contextualizar los resultados obtenidos en función de las tasas de respuesta obtenidas.
A continuación se resaltan algunos aspectos importantes a considerar sobre el tratamiento con la combinación de LDV/SOF.
Genotipo 1
En los pacientes no cirróticos sin tratamiento previo se ha estudiado la conveniencia de acortar la terapia con LDV/SOF añadiendo o no RBV. Los resultados sugieren que estas estrategias obtienen tasas de respuesta viral sostenida similares a las obtenidas con LDV/SOF 12 semanas. El número de pacientes que recayeron fue muy reducido aunque fueron más frecuentes en las pautas de tratamiento de 8 semanas en comparación con las 12 semanas. Los resultados indican que la adición de RBV no incrementa las tasas de RVS12. De forma general, en pacientes naive no cirróticos, el régimen recomendado debe es LDV/SOF 8 semanas. En el contexto de un paciente con bajo riesgo de progresión de la enfermedad y con opciones de retratamiento puede considerarse optar por un régimen respuesta (genotipo IL28 CC y /o carga viral basal (<6 millones UI/ml), decidiendo caso por caso.
Aunque en el análisis de subgrupos del estudio ION3 no se apreciaron diferencias entre los genotipos 1a y 1b, el escaso número de pacientes con genotipo 1b hace que deban mantenerse cautela a la hora de extraer conclusiones.
En los pacientes no cirróticos previamente tratados (con biterapia o triple terapia con un IP) la recomendación general es prolongar la terapia con LDV/SOF durante 12 semanas. Por otro lado, en el caso de que el fallo o la recaída del tratamiento previo incluya fracaso con antivirales de acción directa, habría que considerar una actitud más conservadora que prolongue la terapia a 24 semanas o 12 semanas de LDV/SOF+ RBV, ya que en estos pacientes, pese a no ser cirróticos, las opciones de retratamiento son limitadas. En los pacientes con cirrosis compensada que no han recibido tratamiento previo, la administración de LDV/SOF durante 12 semanas ha mostrado altas tasas de RVS y además se ha observado que la adición de RBV no aporta un aumento en dichas tasas.
En los pacientes con cirrosis compensada tratados previamente (incluidos pacientes con fracaso a triple terapia con IP), la administración de LDV/SOF + RBV durante 12 semanas ha mostrado altas tasas de RVS (96%) y además se ha observado que la prolongar el tratamiento a 24 semanas no aporta un aumento en dichas tasas.
En los pacientes cirróticos descompensados los datos preliminares del estudio SOLAR-1 muestran que la adición de RBV a la terapia con LDV/SOF 12 semanas podría aportar mayores tasas de RVS. También se ha observado en el estudio SOLAR-1 que el aumento de la duración del tratamiento a 24 semanas no aporta mayor eficacia con respecto a la duración de 12 semanas.
Genotipo 2
La eficacia de LDV/SOF en pacientes infectados por el VHC genotipo 2 no ha sido evaluada, por lo que estos pacientes no deben tratarse con LDV en combinación con SOF.
Genotipo 3
La evidencia sobre la eficacia de LDV/SOF durante 12 semanas en pacientes infectados por VHC genotipo 3 es escasa y limitada a poblaciones no muy grandes de pacientes.
SOF + RBV durante 24 semanas, no se ha comparado a LDV/SOF + RBV 12 semanas ni con DCV +SOF 12 semanas.
Mientras no se tengan más datos, la adición de LDV a un régimen de SOF + RBV aumenta la RVS en los pacientes en los que el régimen de SOF + RBV solo no es tratamiento óptimo. En pacientes con infección por VHC genotipo 3 y fallo terapéutico previo y/o pacientes cirróticos, que son aquellos con pocas alternativas terapéuticas, se recomienda el uso de la LDV/SOF + RBV (en vez de SOF + RBV) durante 24 semanas. Hay que resaltar que la duración de 24 semanas no ha sido ensayada en este tipo de pacientes (en el estudio ELECTRON-2 los pacientes fueron tratados durante 12 semanas). La autorización del tratamiento durante 24 semanas en vez de durante 12, viene motivada fundamentalmente por los resultados del estudio VALENCE con SOF + RBV en el que la duración de 24 semanas fue superior a la de 12 y 16, sin embargo esto no ha sido probado con LDV/SOF + RBV. No obstante, en pacientes con genotipo 3 pretratados sin cirrosis, el régimen DCV + SOF durante 12 semanas (estudio ALLY-3) obtiene tasas de RVS del 94%. En pacientes pretratados con cirrosis, este régimen resulta subóptimo (tasa de RVS del 69%) y se recomienda prolongar a 24 semanas la duración del tratamiento. Los otros regímenes sin interferon disponibles hasta la fecha también resultan subóptimos (SOF+RBV 24 semana obtiene tasas de RVS del 60% en pacientes pretratados obtuvo un 83% (20/24) de RVS12 tanto en cirróticos como en no cirróticos.
Genotipo 4
La recomendación de tratamiento con LDV/SOF durante 12 semanas para los pacientes infectados por VHC genotipo 4 se apoya en la actividad antiviral in vitro de ambos compuestos. Los datos de los estudios en los que se ha incluido pacientes con este genotipo parecen apoyar que los datos de eficacia sean similares al genotipo 1.
Esta extrapolación ya ha sido aceptada previamente por el CHMP. Por otra parte los datos de seguridad no se modifican según el genotipo viral. A falta de estimaciones precisas de eficacia, el tratamiento de LDV/SOF debe tener una duración de al menos 12 semanas y hasta 24 semanas según los condicionantes de cada paciente.
LDV/SOF en pacientes coinfectados VIH/VHC
La coinfección con el VIH no afecta negativamente a la actividad de LDV/SOF, aunque el fármaco ha sido ensayado solo en genotipo 1 y en un número muy limitado de pacientes con genotipo 4 (7 pacientes).
LDV/SOF en pacientes con Hepatitis C recurrente post- trasplante hepático
En los pacientes con hepatitis C recurrente post-TH del estudio SOLAR-1, las tasas de RVS12 con LDV/SOF + RBV fueron muy elevadas en pacientes con fibrosis F0-F3 (96-98%) y en aquellos con cirrosis compensada (Child Pugh A - 96%); La tasa de RVS12 en pacientes con cirrosis descompensada fue menor (Child Pugh B→ 83-85%; Child Pugh C→ 60-67%), aunque los porcentajes en los pacientes con cirrosis en estadio Child-Pugh C deben interpretarse con precaución debido al escaso número de pacientes en cada brazo de tratamiento. En general, la prolongación del tratamiento a 24 semanas no supone un incremento en las tasas de RVS12, aunque el número de pacientes en cada subgrupo es pequeño. Estas tasas son similares a las alcanzadas con otros regímenes sin interferón en pacientes con enfermedad menos avanzada. El análisis intermedio del estudio CORAL-I indica que la combinación de ombitasvir/paritaprevir/ritonavir + dasabuvir + RBV durante 24 semanas proporciona tasas de RVS12 similares (97%) en pacientes G1 sin fibrosis o con fibrosis F1-F2. En un estudio reciente de vida real, la combinación SMV + SOF +/- RBV durante 12 semanas consiguió una tasa de RVS12 del 90% en pacientes con genotipo 1, 30% de los cuales tenían enfermedad avanzada (F3-F4) y 11% recidiva colestásica. En pacientes con genotipo 1a, la tasa de RVS12 fue significativamente inferior en los pacientes con F3-F4 (71%) frente a aquellos con F0-F2 (91%), no existiendo esas diferencias en los pacientes con genotipo 1b (34). Datos preliminares de los estudios de vida real de la combinación DCV+SOF +/- RBV 24 semanas en pacientes con recurrencia post trasplante grave proporcionan tasas de RVS12 aproximadamente del 75% (35). Datos de la cohorte CUPILT de 30 pacientes con recurrencia grave (hepatitis colestásica fibrosante) tratados con DCV+SOF 24 semanas muestran tasas RVS12 en los pacientes que ya han finalizado el seguimiento a las doce semanas del 100% (11/11) (36). LDV/SOF tiene, al igual que DCV + SOF, la ventaja de que puede utilizarse en pacientes con cirrosis descompensada (Child-Pugh B y Child-Pugh C) en los que simeprevir ó ombitasvir/paritaprevir/ritonavir no pueden o no deben utilizarse y en tener actividad frente al genotipo 3.
Seguridad
Los datos de seguridad para SOF / LDV proceden de alrededor de 2.000 pacientes. El perfil de seguridad es favorable, con muy pocas interrupciones del tratamiento. SOF / LDV se asocia con un aumento de la frecuencia de dolor de cabeza y de fatiga, que fueron de intensidad leve-moderada. No se han observado otros efectos secundarios con esta combinación de fármacos. Además, en combinación con ribavirina, se observan los efectos secundarios típicos de la RBV (por ejemplo, anemia), pero ésta fue generalmente bien tolerada cuando se utilizó en combinación con SOF / LDV
CONCLUSIONES
LDV/SOF es la primera combinación a dosis fijas de 2 antivirales de acción directa; LDV, un inhibidor específico de la proteína NS5A y SOF, un inhibidor no nucleósido de la polimerasa NS5B.
En pacientes naive y pretratados que sean candidatos al tratamiento con regímenes libres de interferón, LDV/SOF representa una alternativa terapéutica a las otras combinaciones de antivirales de acción directa ya autorizados para el genotipo 1 y 4 del VHC. En pacientes seleccionados (con carga viral<6 millones de UI/ml) también presenta la ventaja de poder acortar la duración de tratamiento (8 semanas en pacientes genotipo 1 naive no cirróticos).
La coinfección con el VIH no afecta negativamente a la actividad de LDV/SOF.
Según los datos del estudio SOLAR-1, LDV/SOF+RBV 12 semanas es una alternativa terapéutica eficaz y segura en pacientes con hepatitis C cirróticos descompensados inelegibles para tratamientos basados en IFN.
LDV/SOF no está indicado en pacientes con genotipo 2.
En pacientes con genotipo 3 los datos de LDV/SOF son muy limitados. En pacientes pretratados sin cirrosis, se considera que el régimen sin interferón preferente en estos momentos es DCV + SOF durante 12 semanas. En pacientes con genotipo 3 pretratados con cirrosis se desconoce la duración óptima de tratamiento de LDV/SOF; los resultados apuntan a considerar que la combinación LDV/SOF + RBV 24 semanas puede aumentar la RVS que se obtiene con SOF + RBV 24 semanas. Sin embargo, esto no se ha demostrado. En estos pacientes, LDV/SOF 24 semanas representaría una alternativa terapéutica a otros regímenes sin interferon (SOF+RBV 24 semanas ó SOF/DCV 24 semanas). Aunque no se han realizado comparaciones directas, desde el punto de vista clínico ninguna combinación libre de IFN (SOF+DCV 12 semanas, SOF+RBV 24 semanas) parece ser superior a SOF + PEG+RBV 12 semanas, por lo que SOF + PEG/RBV durante 12 semanas es la pauta preferente en estos momentos siempre que los pacientes sean susceptibles de ser tratados con IFN.
LDV/SOF + RBV es una alternativa terapéutica eficaz y segura en pacientes con hepatitis C recurrente post-trasplante. En pacientes sin cirrosis (F0-F3), en aquellos con cirrosis compensada (Child- Pugh A) y, posiblemente, en los pacientes con cirrosis descompensada en estadio Child-Pugh B, la prolongación del tratamiento de 12 a 24 semanas no supone un incremento en la tasa de RVS12. El escaso número de pacientes con estadio Child-Pugh C en el estudio SOLAR-1 no permite establecer conclusiones sobre el tiempo óptimo de tratamiento con LDV/SOF + RBV (12 vs 24 semanas) en este subgrupo.
REFERENCIAS
1. Kohli A, Shaffer A. Treatment of hepatitis C: a systematic review. JAMA. 2014 Aug 13;312(6):631-40.
2.Ficha Técnica Interferón disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/000281/WC500034679.pdf
3. Ficha técnica de Boceprevir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002332/WC500109786.pdf
4. EPAR de Boceprevir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002332/WC500109789.pdf
5. Ficha técnica de Telaprevir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf
6. EPAR de Telaprevir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002313/WC500115532.pdf
7. Ficha Técnica de Sofosbuvir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/002798/WC500160597.pdf
8. EPAR de Sofosbuvir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002798/WC500160600.pdf
9. Ficha técnica Daclatasvir disponible en: http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/003768/WC500172848.pdf
10. EPAR de Daclatasvir disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003768/WC500172849.pdf
11. Plan Estratégico Nacional para el Abordaje de la Hepatitis C. Ministerio de Sanidad, Servicios Sociales e Igualdad 12. European Association for the Study of the Liver (EASL). Recommendations on Treatment of Hepatitis C. 2014. Disponible en: http://files.easl.eu/easl-recommendations-on-treatment-of-hepatitis-C/index.html. Último acceso: diciembre de 2014.
13. Guidelines for the screening, care and treatment of persons with hepatitis infection. WHO April 2014. Disponible en: http://apps.who.int/iris/bitstream/10665/111747/1/9789241548755_eng.pdf?ua=1&ua=1
14. Infectious Diseases Society of America (IDSA), American Association for the Study of Liver Diseases (AASLD) and the International Antiviral Society–USA (IAS–USA). Recommendations for Testing, Managing, and Treating Hepatitis C. Disponible en: http://www.hcvguidelines.org/fullreport
15. Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(4):1433-1444.
16. Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335-1374.
17. KDIGO. KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl. 2008;(109):S1-99.
18. Ficha técnica de Harvoni® disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003850/WC500177995.pdf
19. EPAR de Harvoni® disponible en: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003850/WC500177996.pdf
20. Link JO, Taylor JG. Discovery of ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J Med Chem. 2014 Mar 13;57(5):2033-46.
21. Gane EJ, Stedman CA. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology. 2014 Mar;146(3):736- 743.e1.
22. Lawitz E, Poordad FF. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014 Feb 8;383(9916):515-23.
23. Kowdley KV, Gordon SC. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014 May 15;370(20):1879-88.
24. Afdhal N, Zeuzem S. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014 May 15;370(20):1889-98.
25. Afdhal N, Reddy KR. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014 Apr 17;370(16):1483-93.
26. Bourliere M, Bronowicki JP, Omata M et al. An Integrated Safety and Efficacy Analysis of >500 Patients With Compensated Cirrhosis Treated With Ledipasvir/Sofosbuvir With or Without Ribavirin. Oral communication presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA.
27. Gane EJ, Hyland RH, An D, et al. Sofosbuvir/ledipasvir fixed dose combination is safe and effective in difficult-to-treat populations including genotype-3 patients, decompensated genotype-1 patients, and genotype-1 patients with prior sofosbuvir treatment experience. Program and abstracts of the 49th Annual Meeting of the European Association for the Study of the Liver; April 9-13, 2014; London, England. Abstract 06.
28. Gane EJ, Hyland RH, An D, et al. High Efficacy of LDV/SOF Regimens for 12 Weeks for Patients With HCV Genotype 3 or 6 Infection. Poster presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA..
29. Flamm SL, Everson GT, Charlton M, et al. LDV/SOF +RBV for the Treatment of HCV in Patients with Decompensated Cirrhosis: Preliminary Results of a Prospective, Multicenter Study. Oral communication #239 presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA.
30. Reddy R. LDV/SOF + RBV for the Treatment of HCV in Patients with Post Transplant Recurrence: Preliminary Results of a Prospective, Multicenter Study. Oral communication #8 presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA.
31. Bourliere M, Bronowicki JP, de Ledinghen V, et al. Ledipasvir/Sofosbuvir Fixed-Dose Combination Is Safe and Efficacious in Cirrhotic Patients Who Have Previously Failed #LB-6 presented at AASLD: The Liver Meeting®, November 7–11, 2014, Boston, MA.
32. Naggie S, Cooper C, Saag M, et al. Ledipasvir/sofosbuvir for 12 Weeks in Patients Coinfected With HCV and HIV-1: ION-4. 2015 Conference on Retroviruses and Opportunistic Infections. Seattle, February 23-24, 2015. Abstract 152LB
33. Osinusi A, Townsend K, Nelson A, et al. Use of sofosbuvir/ledipasvir fixed dose combination for treatment of HCV genotype-1 in patients coinfected with HIV. Journal of the American Medical Association. February 23, 2015 (Epub ahead of print).
34. Pungpapong S, et al. Multicenter experience using sofosbuvir and simeprevir with/without ribavirin to treat HCVgenotype 1 after liver transplantation. American Association for the Study of Liver Diseases (AASLD) Liver Meeting. Boston, November 7-12, 2014; Abstract 9.
35. Fontana R, Bahirwani R, Reddy R, et al. High Efficacy and Favorable Safety Profile of Daclatasvir Based All Oral Antiviral Therapy in Liver Transplant Recipients with Severe Recurrent HCV. American Association for the Study of Liver Diseases (AASLD) Liver Meeting. Boston, November 7-12, 2014. AbstractLB-22.
36. Leroy V, Dumortier J, Coilly A, et al. High Rates Of Virological Response And Major Clinical Improvement during Sofosbuvir and Daclatasvir-based Regimens for the Treatment of Fibrosing Cholestatic HCV-Recurrence after Liver Transplantation: The ANRS CO23 CUPILT Study. American Association for the Study of Liver Diseases (AASLD) Liver Meeting. Boston, November 7-12, 2014. Abstract21
GRUPO DE EXPERTOS
(por orden alfabético)
Agencia Española de Medicamentos y Productos Sanitarios
Centro de Información Farmacoterapéutica del Servizo Galego de Saúde
Comité de Evaluación de Nuevos Medicamentos en el Ámbito Hospitalario de Euskadi
Comunidad Autónoma de Andalucía
Grupo Técnico de Utilidad de Medicamentos Hospitalarios de la Comunidad de Madrid
Arantxa Sancho López
Servicio de Farmacología Clínica. Hospital Universitario Puerta de Hierro Majadahonda, Madrid. Miembro del Comité de Medicamentos de Uso Humano (CHMP) de la EMA.
Todos los expertos han realizado una declaración de conflictos de interés.
El Laboratorio Titular, la Sociedad Española de Trasplante Hepático, Sociedad Española de Farmacología Clínica, Sociedad Española de Farmacia Hospitalaria, Asociación Española para el Estudio del Hígado, Federación Nacional de Enfermos y Trasplantados Hepáticos, la Federación Española de Hemofilia y el Foro Español de Activistas en Tratamientos del VIH han tenido oportunidad de enviar comentarios al documento, si bien el texto final es el adoptado por el GCPT.
Fuente: Agencia Española de Medicamentos y Productos Sanitarios.
