Informe de Posicionamiento Terapéutico de efmoroctocog alfa (Elocta®) en hemofilia A
INFORME DE POSICIONAMIENTO TERAPÉUTICO
Informe de Posicionamiento Terapéutico de efmoroctocog alfa (Elocta®) en hemofilia A
IPT, 51/2016
Versión 1
Fecha de publicación: 21 de noviembre de 2016 †
El factor VIII de coagulación (FVIII) es una glicoproteína que circula en el plasma sanguíneo formando un complejo mediante un enlace no covalente, esencial para conseguir una hemostasia adecuada. El defecto congénito de esta proteína da lugar a la enfermedad hereditaria, hemofilia A (HA).
Los pacientes con HA tienen un riesgo hemorrágico importante debido a la deficiencia congénita del FVIII, causada por una mutación en el gen F8 localizado en el brazo largo del cromosoma X. La herencia va ligada al sexo de tal manera que las mujeres son portadoras y los hombres padecen la enfermedad.
De acuerdo a su severidad, la hemofilia A se subdivide en grave, moderada y leve. La grave (nivel de FVIII <1UI/dL) comprende un 32,3% (1) y se manifiesta clínicamente por la propensión a sufrir episodios de sangrado espontáneos que a menudo afectan a las articulaciones (fundamentalmente tobillos, hombros y rodillas), y sangrados en los músculos, tejidos blandos, tracto gastrointestinal y Sistema Nervioso Central (SNC). Los sangrados recurrentes en las articulaciones pueden desencadenar una artropatía crónica, causando una gran morbilidad a los pacientes. Los pacientes con HA moderada (nivel de FVIII 1-5 UI/dL) sangran a consecuencia de heridas o traumatismos de pequeña cuantía y también pueden desarrollar una artropatía hemofílica. Los pacientes con HA leve (nivel de FVIII >5-<40 UI/dL) sangran generalmente tras traumaumatismos importantes o tras cirugías (2).
Según los datos de la Federación Mundial de Hemofilia (3), la población hemofílica en todo el mundo es alrededor de 142.205 personas, lo que supone una prevalencia de 0,2 casos por 10.000 personas. En la Unión Europea la prevalencia es de 0,6 casos por 10.000 personas. En España el número de pacientes con HA es de 2.595 (4).
Actualmente no existe un tratamiento curativo para la HA, y el enfoque terapéutico consiste en la reposición del factor deficiente utilizando medicamentos con FVIII obtenido de plasma (FVIIIp) o con FVIII recombinante (FVIIIr). La utilización de estos medicamentos puede ser "a demanda" para parar un episodio de sangrado manifiesto, en profilaxis previa a una cirugía o como profilaxis con administraciones regulares para prevenir los sangrados.
El abordaje más recomendado es el tratamiento profiláctico. Dado que la vida media (t 1/2 ) de la mayoría de los concentrados de FVIII es de 12 horas, este régimen supone administraciones intravenosas frecuentes (3 ó 4 veces por semana) resultando un tratamiento muy gravoso para el paciente, condicionando su calidad de vida, y siendo un problema importante la adherencia al tratamiento (5).
EFMOROCTOCOG ALFA (ELOCTA ®)
Es una proteína de fusión de factor VIII recombinante con deplección del dominio B, unido covalentemente a la fracción constante (Fc) de la inmunoglobulina humana (IgG1). Consta de 1.890 aminoácidos y es producido por tecnología recombinate en células embrionarias de riñón humano (HEK) sin adición externa de proteínas animales o humanas durante el proceso de fabricación.
Se presenta como un vial polvo (250 UI, 500 UI, 750 UI, 1.000UI, 1.500 UI, 2.000UI y 3.000UI) y un disolvente para solución inyectable en jeringa precargada.
Está indicado para el tratamiento y la profilaxis de sangrado en pacientes con HA (causada por la deficiencia congénita de FVIII).
Farmacología
Cuando se administra mediante inyección intravenosa en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/ factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo.
La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
La región Fc de la inmunoglobulina (Ig) se une al receptor neonatal para la Fc, este receptor se expresa durante toda la vida y es parte del proceso fisiológico por el cual las inmunoglobulinas se protegen de la degradación lisosomal proporcionando una vida media larga a estas proteínas. Por tanto, la estructura molecular de efmoroctocog alfa está diseñada para alargar su t 1/2 .
Los principales datos de PK derivan del ensayo clínico fase I (998HA101). En él se estudió el comportamiento PK de dos dosis de efmoroctocog alfa (25 y 65 UI/Kg) y se comparó con octocog alfa. Los resultados de este estudio se consideran muy relevantes puesto que hablando de factores de coagulación la PK es un variable subrogada de la eficacia del medicamento.
La recuperación de octocog alfa y efmoroctocog alfa fue muy parecida, 55,09 IU/dl y 106,59 IU/dl para 25 y 65 IU/Kg en el caso de efmoroctocog alfa y 56,36 IU/dl y 116,52IU/dl para 25 IU/kg y 65 IU/kg en octocog alfa.
Sin embargo se apreciaron diferencias en cuanto a la vida media siendo 17,66 h y 17,24 h para 25 IU/Kg y 65IU/Kg en el caso de efmoroctocog alfa, y 11,56 h y 9,79 h para 2.5IU/kg y 65IU/kg en el caso de octocog alfa. Esto supone una prolongación de la t 1/2 para efmoroctocog alfa de: 1,53 veces (95% C.I; 1,36 – 1,72; p=0.0002) en 25 IU/kg y una prolongación de 1,76 veces (95% C.I; 1,57 – 1,98; p<0,0001) en la dosis de 65 IU/kg.
La Compañía también realizó otras investigaciones del comportamiento PK de efmoroctocog alfa en los estudios fase II y III, y un estudio de PK poblacional. Los datos obtenidos en cada uno de ellos corroboran el efecto de larga duración de efmoroctocog alfa debido a una t 1/2 más prolongada. De manera que el tiempo transcurrido hasta que la actividad declina a 1 UI/dl es alrededor de 4,9 días en efmoroctocog alfa y 3,2 días en octocog alfa. Como se ha observado con otros factores VIII el aclaramiento es más rápido en los niños menores de 6 años (3,86 ml/h/Kg) comparado con los niños más mayores (3,05 ml/h/Kg).
Los resultados del modelo de PK poblacional mostraron que el 90% de los pacientes de 12 años o más, tendrían niveles superiores al 1% con un régimen de 50 UI/kg cada 3 días, la proporción descendería al 64,5% y 42,6% si las dosis fueran administradas cada 4 o 5 días.
En niños de 6 años, una dosis de 50 IU/kg cada tres días cubriría al 79,5% de la población (con niveles valle superiores al 1%), si el intervalo se prolongara a 4 días el porcentaje descendería al 43,1%.
Eficacia
La eficacia de efmoroctocog alfa se ha estudiado en un ensayo clínico confirmatorio fase III (997HA301) en pacientes adultos y adolescentes mayores de 12 años, en un estudio en población pediátrica (8HA02PED) con niños menores de 12 años y en estudio a largo plazo para investigar fundamentalmente la seguridad.
Todos los pacientes incluídos en los ensayos clínicos eran previamente tratados (PPT) con más de 150 días de exposición (DE) a tratamientos previos en la población adulta y más de 50 DE en los niños. Todos los pacientes tenían HA grave con un nivel de factor <1 UI/dl o tenían un genotipo indicativo de HA grave. Durante el desarrollo solamente 2 pacientes mayores de 65 años fueron reclutados.
Fueron sistemáticamente excluidos todos los pacientes con inhibidores, con otras alteraciones de la coagulación diferentes a la HA, o con historia de hipersensibilidad al FVIII o a la administración intravenosa de Ig.
Estudio 997HA301(6)
Se trata de un estudio fase 3, abierto, muticéntrico con tres brazos de tratamiento: el brazo 1 consiste en un profilaxis individualizada y ajustada al paciente (según sus datos de PK) para lograr un nivel valle entre el 1% al 3%, el brazo 2 consiste en un régimen semanal de profilaxis y el brazo 3 consiste en un brazo a demanda (ver figura 1).
Figura1. Esquema del diseño del estudio fase III 997HA301
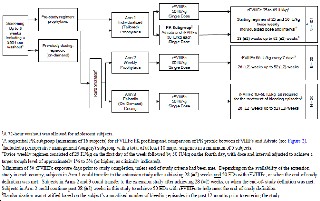
Los tratamientos consistieron en: brazo 1 dosis inicial de 25 UI/Kg en el día 1, y 50 UI/Kg en el día 4. Un subgrupo de pacientes del brazo 1 fueron incluidos en un estudio de PK en el cual los parámetros PK de efmoroctocog alfa fueron comparados con octocog alfa. En el brazo 2 el régimen prescrito fue 65 UI/kg administradas cada 7 días; y en el brazo 3, dosis entre 10-50 UI/kg como fuese necesario para parar el sangrado.
Hubo un subgrupo de pacientes que requirió profilaxis debido a un procedimiento quirúrgico.
Todos los pacientes con profilaxis previa a la entrada en el estudio entraron en el brazo 1, y los pacientes a demanda antes de entrar en el estudio podían ser aleatorizados al brazo 2 o 3.
Ciento sesenta y cinco pacientes fueron reclutados en 75 centros: 118 en el brazo 1, 24 en el brazo 2 y 23 en el brazo 3. El análisis de PK se estudió en 30 pacientes y 9 fueron sometidos a cirugía.
El 68,5% de los pacientes tuvieron una articulación diana con artropatía a nivel basal (61,9% en el brazo 1, 91,7% en el brazo 2 y 78,3% en el brazo 3).
Estudio 8HA02PED (7)
Se trata de un estudio abierto, no aleatorizado. El estudio incluye una cohorte de niños menores de 6 años y otra de niños hasta los 12 años de edad. En todos ellos se llevó a cabo una evaluación PK de efmoroctocog alfa y se comparó con los datos de PK del factor FVIII administrado previo al inicio del estudio.
El régimen profiláctico consistía de 25 UI/kg el día 1 y 50 UI/Kg el día 4, se permitía un ajuste de dosis en el rango de 25-80 UI/kg y un ajuste del intervalo de dosis con un mínimo de 2 días de separación entre administraciones, con el objetivo de mantener un nivel valle de actividad del 1%.
Un total de 71 pacientes fueron reclutados, 69 fueron tratados con efmoroctocog alfa (35 menores de 6 años y 34 entre 6 y 12 años) y 60 fueron incluidos en la PK. Siete pacientes tuvieron procesos quirúrgicos menores.
El 18,3% de los niños tenían una articulación diana.
Estudio 8HA01EXT(8)
El objetivo de este estudio es recabar datos de seguridad de los pacientes por lo menos durante 100 DE o hasta que el medicamento esté comercialmente disponible y obtener datos de eficacia con regímenes de profilaxis más abiertos; permitiéndose en los adultos y adolescentes la pauta de 1) profilaxis individualizada 25-65 UI/kg cada 3-5 días, 2) dos veces a la semana comenzando con 20-65 UI/kg en el día 1 y 40-65 UI/kg en el día 4, 3) profilaxis modificadal o 4) régimen a demanda, y en los niños de hasta 80 UI/Kg con dos días de separación entre dosis.
En cada uno de los estudios se evaluó la eficacia de efmoroctocog alfa en profilaxis, a demanda y en cirugía.
Además la Compañía presentó datos post-hoc del cambio de la tasa anualizada de sangrados (TAS) y las dosis profilácticas antes del estudio (pre-study) comparándolas con los datos durante el estudio (on-study) (9).
Los países con más alta tasa de reclutamiento fueron Estados Unidos, Reino Unido y Sudáfrica.
Eficacia en profilaxis
La variable más importante fue la tasa anualizada de sangrado (TAS).
Estudio 997HA301
La TAS fue 2,91 (95% CI, 2,30- 3,68) en el brazo 1, 8,92 (95% CI, 5,48- 14,51) en el brazo 2, y 37,25 (95% CI, 24,03- 57,74) en el brazo 3. Los valores de la mediana y rango intercuartílico (RIC) fueron 1,60 (0,0; 4,7); 3,89 (1,86; 8,36); 33,57 (21,14; 48,69) para los brazos 1, 2, y 3, respectivamente.
El porcentaje de pacientes sin ningún episodio de sangrado fue más pequeño en el brazo 2 (17,4%) que en el brazo 1 (45,3%).
En el brazo 2, el 20,8% de los pacientes discontinuó el tratamiento prematuramente.
Para la mayoría de los pacientes en el brazo 1 (82/117) el intervalo de dosificación fue 3-5 días, siendo la media de dosis profiláctica 50,41 UI/Kg cada 3 días en 40 pacientes, 47,85 UI/kg cada 4 días en 3 pacientes y 50,61 UI/Kg cada 5 días en 29 pacientes. La media de los días transcurridos entre dosis fue 3,51 (rango 2,9-5,0 días).
La mediana del número de inyecciones mensuales fue 8,88 (7,27- 9,87), 4,98 (4,67- 5,57) y 3,25 (2,05- 4,94) en los brazos 1, 2 y 3 respectivamente.
La media (desviación estándar) del consumo mensual y anual en UI/kg fue 386,00 (86,801) y 4.631,98 (1041,608) en el brazo 1, 333,64 (54,381) y 4.003,69 (652,573) en el brazo 2, y 108,70 (72,863) y 1.304,36 (874,361) en el brazo 3.
Estudio 8HA02PED
La mediana de la TAS fue 0,00 con un rango interquartílico (RIC) de 0,0 a 3,96 en los niños menores de 6 años, y 2,01 (RIC de 0,0 a 4,04) en los niños de 6 a 12 años.
Los valores medianos de TAS fueron 1,96 (RIC, 0,0- 3,96). El valor medio de TAS fue 2,25 en los <6 años y 2,99 en los más mayores.
Treinta y dos pacientes (46,4%) no tuvieron ningún evento.
La mediana de la dosis profiláctica semanal fue 91,63 IU/kg (RIC: 84,72; 104,56 IU/kg) y 86,88 IU/kg (RIC: 79,12; 103,08 IU/kg) en cada cohorte de edad.
El intervalo entre inyecciones fue 3,49 días (RIC: 3,46; 3,51 días), en ambas cohortes.
El número de inyecciones mensuales fue 9,14 (8,87- 9,49) en los <6 años y 9,03 (8,88- 9,25) en los niños de 6-12 años.
La media (desviación estándar) del consumo mensual y anual fue 444,32 (92,233) y 5.331(1106,677) en niños menores de 6 años y 414,46 (81,338) y 4.973,54 (976,060) en niños entre 6 y 12 años.
Solamente 15 de 69 pacientes estuvieron en un tratamiento regular, la mayoría (54/69) 78,3% fueron tratados con un régimen irregular con dosis alternantes, siendo la mediana más baja 31,73 UI/Kg y la más alta 55,87 UI/Kg.
Eficacia en el control del sangrado
Las variables más importantes fueron: la respuesta hemostática según una escala de cuatro puntos (excelente, buena, moderada, o no respuesta) después de la primera inyección (Tabla 1); el consumo por inyección y el consumo por evento.
Tabla 1: Escala para la evaluación hemostática de los episodios de sangrado
Estudio 997HA301
Hubo 757 episodios de sangrado (ES), (209 en el brazo 1; 92 en el brazo 2 y 456 en el brazo 3).
La evaluación de la respuesta hemostática fue excelente o buena en el 77,6% de los eventos tras la primera administración. El 87,3% de los ES se resolvieron con una única inyección.
La dosis media administrada para resolver un ES fue 44,53, 36,02 y 32,63UI/Kg en los brazos 1, 2 y 3 respectivamente.
Estudio 8HA02PED
Hubo 86 ES (38 y 48 en cada cohorte de edad).
La respuesta hemostática a la primera inyección fue valorada como excelente o buena en el 89,4% de los casos, moderada en el 8,7% y como ninguna en el 1,9%.
El 18,6% de los ES necesitaron 2 o más inyecciones para ser controlados.
La dosis necesaria para la resolución de un ES fue 54,90 IU/kg. Eficacia en cirugía
La variable más importante fue un análisis de las respuestas hemostáticas realizadas por el cirujano según una escala de cuatro gradientes, la pérdida de sangre y la necesidad de transfusiones.
Durante todo el desarrollo clínico contando con el estudio de extensión, hubo 24 cirugías mayores. La mayoría fueron cirugía ortopédica (siendo la más común la implantación de prótesis de rodilla y cadera). La respuesta hemostática fue valorada como excelente o buena en 22 cirugías mayores y en 32 cirugías menores
El día de la cirugía la mayoría de los pacientes recibieron 2 inyecciones, con una dosis total entre 50,8 y 126,6 IU/kg/día. En los tres primeros días después de la operación el rango de dosis fue 15,3–79,1 IU/kg/día. Desde el día 4 al día 14 después de la operación los pacientes recibieron entre 1 y 22 inyecciones con un rango de 9,4 a 66,3 IU/kg/día.
Tres pacientes precisaron transfusión de concentrado de hematíes.
Los datos post-hoc que presentó la Compañía comparando la dosis y los sangrados pre-estudio y on-estudio mostraron un descenso similar en la TAS y una disminución del número de inyecciones entre 40-100 al año para los pacientes adultos y adolescentes, y entre el 40-80 en los niños, mientras el consumo de factor era similar. El problema es que estos datos fueron recopilados retrospectivamente, y para el cálculo de la dosis pre-studio no se disponía de una recogida sistemática de datos, de manera que los resultados son susceptibles de sesgos.
Seguridad
La seguridad de efmoroctocog alfa se ha estudiado en 233 pacientes, la mayoría ha estado expuesta a efmoroctocog alfa durante más de 50DE (sólo el 7,7% tuvo un exposición menor).
Treinta y nueve pacientes (16,7%) tuvieron 55 eventos adversos graves, ninguno relacionado con efmoroctocog alfa.
Dos de estos eventos ocurrieron en un paciente que falleció por consumo de estupefacientes y suicidio.
Los eventos de especial interés que se produjeron fueron 12 reacciones alérgicas no graves y 4 eventos tromboembólicos no graves, ninguno relacionado con efmoroctocog alfa.
De los 22 pacientes sometidos a cirugía mayor, 8 tuvieron 18 acontecimientos adversos graves durante el periodo post-cirugía; el más frecuente fue fiebre (3 casos), anemia (2 casos) y una hemorragia postprocedimiento.
Se detectaron 25 reacciones adversas relacionadas con efmoroctocog alfa que aparecen recogidas en la FT, la incidencia fue muy baja (0,4%).
Durante el desarrollo clínico 5 pacientes abandonaron el tratamiento con efmoroctocog alfa debido las siguientes reacciones adversas: artralgia, rash, fractura de fémur, abuso de estupefacientes y elevación de creatinina.
Un paciente tuvo un título bajo de anticuerpos neutralizantes (0,73 BU/mL) en una única ocasión y no se pudo confirmarse en análisis repetidos.
Además 12 pacientes tuvieron anticuerpos no neutralizantes (binding antibodies) antes el inicio del tratamiento, en un paciente además con PK alterada del FVIII (mayor aclaramiento y recuperación). Otros 6 pacientes desarrollaron anticuerpos no neutralizantes durante el tratamiento uno de estos pacientes fue el que desarrolló anticuerpos neutralizantes.
Efmoroctocog fue aprobado en Estados Unidos en junio de 2014. La experiencia postcomercialización recoge la ocurrencia de un caso de anticuerpos inhibidores con título alto (37 BU/mL en el primer análisis y 27 BU/mL en el test repetido) en un paciente de 11 meses mínimamente tratado.
Aunque hay un riesgo teórico de que efmoroctocog alfa al llevar una fracción Fc pudiera estar asociada con una mayor incidencia de infecciones (aumento del catabolismo de las Igs por saturación del receptor) esto no ha ocurrido durante los estudios, y no se ha detectado una alteración de los niveles de Ig ni un aumento de la incidencia de infecciones en la población estudiada.
>De manera que en términos generales efmoroctocog alfa es bien tolerado y el perfil de seguridad es el conocido para otros FVIII, sin la ocurrencia de señales inesperadas.
DISCUSIÓN
El desarrollo clínico de efmoroctocog alfa es acorde a los requisitos de las guías europeas respecto al número de pacientes estudiados y el tiempo de seguimiento (10-13).
A pesar de ser una enfermedad rara se dispone de datos de seguridad en 233 pacientes, y el tiempo de seguimiento superó los 50DE en la mayoría de los pacientes, lo cual se considera suficiente para estudiar el riesgo de anticuerpos neutralizantes en la fase preautorización.
Efmoroctocog alfa ha demostrado ser eficaz y seguro en la profilaxis de ES, en el tratamiento de eventos y en el contexto clínico de la cirugía.
Las variables analizadas muestran una TAS y un consumo bastante parecido al de los otros factores (5, 14, 15, 16, 17).
Efmoroctocog alfa es el primer FVIII autorizado con una vida media más prolongada comparada al arsenal terapéutico disponible hoy en día. El problema ha sido que los datos obtenidos a nivel de PK no se han traslado de manera generalizada en una prolongación de los intervalos de dosificación.
La ventaja observada de una t 1/2 1,53 veces más prolongada que octocog alfa no se ha traducido en un efecto directo de prolongar el intervalo de dosificación en esa magnitud, de manera que precisamente el régimen semanal (65 UI/kg cada 7 días) que hubiera supuesto un ganancia relevante para el alivio de la carga de la profilaxis de los paciente, no ha demostrado los datos de eficacia suficientes como para su autorización. Así de los 233 pacientes reclutados, sólo 37 tuvieron esta pauta en algún momento del desarrollo clínico y solo 13 permanecieron en este régimen durante todo el periodo del estudio. Por tanto, dada la escasez de evidencia, ni siquiera se ha aprobado este régimen en ficha técnica (18).
Un intervalo de dosificación >5 días no está avalado por los datos clínicos ni por los datos de PK poblacional y en los niños, pacientes candidatos por excelencia a iniciar profilaxis, ni siquiera se han ensayado intervalos superiores a tres días.
El hecho de que finalmente la pauta posológica en profilaxis sea 50 UI/Kg cada 3 a 5 días, difiere poco de la dosis estándar de 20-40 UI/Kg a intervalos de 2-3 días. La modificación de la molécula de FVIII añadiendo la fracción Fc ha aportado una ventaja PK con una relevancia clínica muy relativa para los pacientes. El hecho de disminuir una inyección a la semana en algunos pacientes se traslada en un menor número de inyecciones durante un año, lo que puede ser llamativo en una enfermedad crónica como la HA. Sin embargo, en los aspectos prácticos, parece que esta pequeña ganancia sería más relevante por ejemplo en pacientes poco cumplidores o en aquellos con necesidad imperiosa de preservar el acceso venoso. Hay que pensar que hoy por hoy no sabemos si efmoroctocog alfa va a proporcionar una mejor adherencia o una mejor calidad de vida.
Los resultados globales indican que la pequeña prolongación del intervalo de dosificación conlleva un incremento de las dosis individuales, aunque el consumo anual permanece similar al de otros medicamentos.
Aunque las dosis individuales sean más altas (el ajuste de dosis permite 65 UI/kg) no se han observado problemas de seguridad ni un incremento en la incidencia de eventos tromboembólicos. Sin embargo, el hecho de que puedan administrar hasta 65 UI/Kg de manera reiterada hace que el riesgo no sea descartable.
Los datos de profilaxis en los niños han mostrado resultados inusuales, ya que pocos niños se mantuvieron en un régimen regular, se desconoce si esto puede tener repercusión en la práctica clínica diaria.
En el tratamiento a demanda de los eventos de sangrado, una ampliación de los intervalos de dosificación cuando se necesitan dosis repetidas no parece justificado, y en principio deben seguirse las pautas estándares para todos los FVIII, que son las establecidas en la FT (12).
El desarrollo clínico no ha incluido pacientes naïve (no tratados previamente), pero un estudio (997HA306) en esta población está en curso tras la autorización de efmoroctocog alfa.
Aunque el perfil de seguridad es aceptable, se han detectado anticuerpos inhibidores en la experiencia postcomercialización fuera de EU. En general, aunque asumimos un perfil de reacciones adversas comparable a los otros medicamentos autorizados, hay que tener en cuenta que los pacientes con hipersensibilidad a Igs no son candidatos a este tratamiento.
CONCLUSIÓN
Efmoroctocog alfa es un medicamento eficaz y seguro en el tratamiento y la profilaxis de la HA tanto en niños como en adultos previamente tratados.
Tiene unas características PK diferentes a los medicamentos autorizados, con una vida media algo más prolongada, pero que no se traducen en un cambio sustancial en los regímenes de tratamiento.
Efmoroctocog alfa proporciona alguna ventaja respecto a un menor número de inyecciones semanales en el régimen de profilaxis. Para el tratamiento a demanda y en cirugía efmoroctocog alfa no proporciona ninguna ventaja respecto al arsenal disponible.
No se dispone de datos clínicos (o estos son limitados) del uso de Efmoroctocog alfa en Inmunotolerancia y en pacientes no tratados previamente.
CONSIDERACIONES FINALES DEL GCPT
La elección entre efmoroctocog alfa y las alternativas se basará fundamentalmente en criterios de eficiencia.
REFERENCIAS
1. Aznar JA, Lucía F, Abad-Franch L Lucía F, Abad-Franch L, Jiménez-Yuste V, Pérez R, Batlle J, Balda I, Parra R, Cortina VR Haemophilia in Spain. Hemophilia 2009; 15:665-75.
2. Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al.
Guidelines for the management of hemophilia. Haemophilia. 2013;19(1): e1-47.
3. WFH Report on the Annual Global Survey 2013. Monteal, Quebec: Word Federation of Heamophilia;2014.
http://www1.wfh.org/publications/files/pdf-1591.pdf. [Acceso Octubre 2015].
4. Hemofilia. Aspectos Organizativos. Ministerio de Sanidad, Servicios Sociales e Igualdad. (noviembre 2012).
http://www.msssi.gob.es/profesionales/saludPublica/medicinaTransfusional/publicaciones/docs/Hemofilia_AspectosOrganizativos.pdf [Acceso Octubre 2015].
5. Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (guardian1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia: 2013;19(5):691-7.
6. Mahlangu J, Powell JS, Ragni VM et al. Phase 3 study of recombinant factor VIII Fc fusión protein in severe hemophilia A. Blood 2014; 123(3): 317-323.
7. Young G, Mahlangu J, Kulkarni R et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia. J of Thromb Haemost 2015; 13: 967–977.
8. Nolan B, Mahlangu J, Perry D et al. Long-term safety and efficacy of recombinant factor VIIIFc fusion protein (rFVIIIFc) in subjects with haemophilia A. Haemophilia 2015;1-9.
9. Shapiro AD et al. Recombinant factor VIII Fc fusion protein: extended interval dosing maintains low bleeding rates and correlates with von Willebrand factor levels. J Thromb Haemost 2014; 12(11): 1788-1800).
10. Guideline on the clinical investigation of recombinant and human plasma-derived factor VIII products EMA/CHMP/BPWP/144533/2009
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109692.pdf. [Acceso Octubre 2015].
11. Workshop report: Characterisation of new clotting factor concentrates (FVIII, FIX) with respect to 587 potency assays used for labelling and testing of post infusion samples, 28-29 November 2013 (EMA/135928/2014)
http://www.ema.europa.eu/docs/en_GB/document_library/Report/2014/07/WC500169760.pdf. [Acceso Octubre 2015].
12. Dodt, J., Hubbard, A. R., Wicks, S. J., Gray, E., Neugebauer, B., Charton, E. and Silvester, G. (2015), Potency determination of factor VIII and factor IX for new product labelling and postinfusion testing: challenges for caregivers and regulators. Haemophilia. doi: 10.1111/hae.12634 595.
13. Guideline on core SmPC for human plasma derived and recombinant coagulation factor IX products EMA/CHMP/BPWP/1625/1999 rev. 2.
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/12/WC500179583.pdf. [Acceso Octubre 2015].
14. Pollmann H, Externest D, Ganser A, et al. Efficacy, safety and tolerability of recombinant factor VIII (REFACTO) in patients with haemophilia A: interim data from a postmarketing surveillance study in Germany and Austria. Haemophilia: 2007;13(2):131-43.
15. Recht M, Nemes L, Matysiak M, et al. Clinical evaluation of moroctocog alfa (AF-CC), a new generation of B-domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia
A: demonstration of safety, efficacy, and pharmacokinetic equivalence to fulllength recombinant factor VIII. Haemophilia: 2009;15(4):869-80.
16. Tarantino MD, Collins PW, Hay CR, et al. Clinical evaluation of an advanced category antihaemophilic factor prepared using a plasma/albumin-free method: pharmacokinetics, efficacy, and safety in previously treated patients with haemophilia A. Haemophilia: 2004;10(5):428-37.
17. Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on-demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10(3):359-67.
18. Elocta®. Ficha técnica autorizada. Disponible en:
http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Product_Information/human/003964/WC500198642.pdf
[Acceso Octubre 2015].
GRUPO DE EXPERTOS
(por orden alfabético)
Agencia Española de Medicamentos y Productos Sanitarios
Todos los expertos han realizado una declaración de conflictos de interés.
El Laboratorio Titular y la Sociedad Española de Farmacología Clínica, la Sociedad Española de Farmacia Hospitalaria, la Sociedad Española de Trombosis y Hemostasia, la Sociedad Española de Hematología y Hemoterapia, y la Federación Española de Hemofilia han tenido oportunidad de enviar comentarios al documento, si bien el texto final es el adoptado por el GCPT.

